十五项耳聋基因芯片联合一代测序检测聋儿家庭的致聋变异分析 |
您所在的位置:网站首页 › 听力基因筛查2168a杂合突变 › 十五项耳聋基因芯片联合一代测序检测聋儿家庭的致聋变异分析 |
十五项耳聋基因芯片联合一代测序检测聋儿家庭的致聋变异分析
|
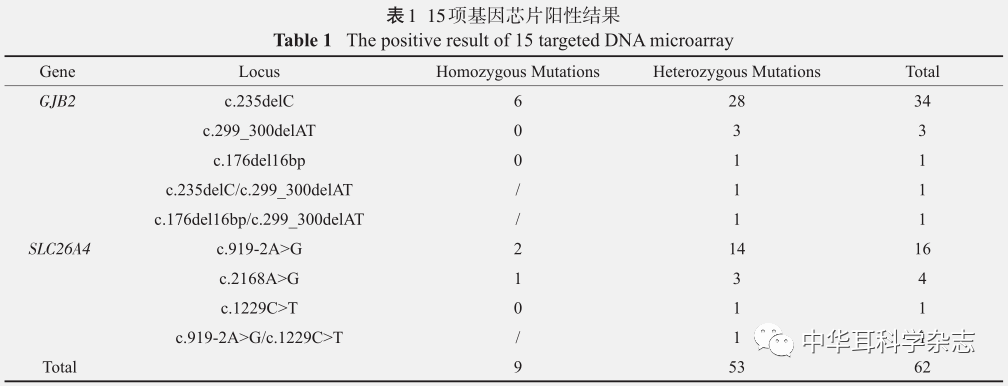
DNA测序结果与NCBI 网站(http://www.ncbi.nlm.nih.gov/)公布的标准序列进行序列比对分析。 2 结 果 2.1 耳聋基因芯片检测结果 在80组先证者家庭中,25组家庭成员携带耳聋相关突变,16组家庭为GJB2基因(占先证者的20%),9组家庭为SLC26A4基因(占先证者的 11.25%),家庭的总检出率为31.25%(25/80)。所有 241人中,GJB2及 SLC26A4基因共 62 例,总检出率为 25.73%(62/241),以GJB2 c.235delC(34/62)与 SLC26A4 c.919-2A>G(16/62)热点变异居多。GJB2基因中,c.235delC纯合变异占总人数的2.49%(6/241),杂合变异占总人数的11.62%(28/241),c.299_300delAT杂合变异占总人数的1.24%(3/241),检测出c.176del16bp杂合变异、c.235delC/c.299_300delAT复合、c.176del16bp/c.299_300delAT复合杂合变异各1例;SLC26A4基因中,c.919-2A>G纯合变异占总人数的0.83%(2/241),杂合变异占总人数的5.81%(14/241),c.2168A>G纯合变异占总人数的0.41%(1/241),杂合变异占总人数的1.24%(3/241),检 测 出c.919-2A>G/c.1229C>T复合杂合变异1例;野生型合计179人,未检测出MT-RNR1及GJB3基因。具体结果如下表(表1)。
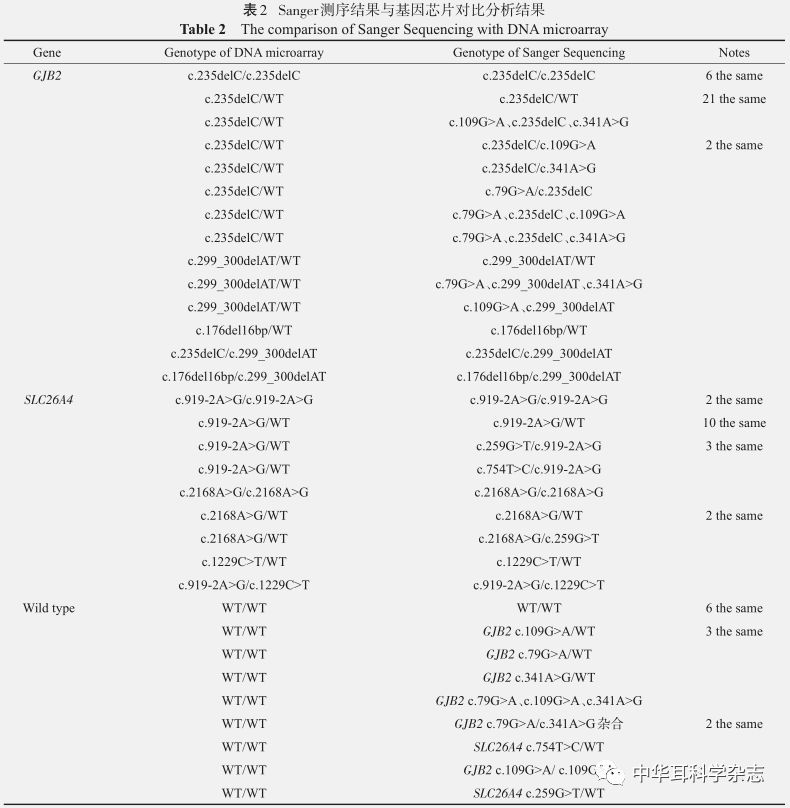
2.2 基因测序结果 对携带基因芯片阳性的25组家庭(包括野生型成员)共79人进行Sanger测序。62例基因芯片阳性结果通过测序验证,符合率达100%;除基因芯片包含的位点外,还检测出部分多态性、点突变等,共计阳性结果为 73 例,总检出率为 30.30%(73/241),其中GJB2 49例,SLC26A4 24例;在进行GJB2基因测序时,发现c.109G>A、c.79G>A、c.341A>G变异;在进行SLC26A4基因测序时,发现 c.259G>T、c.754T>C变异。具体结果如下表(表2)。
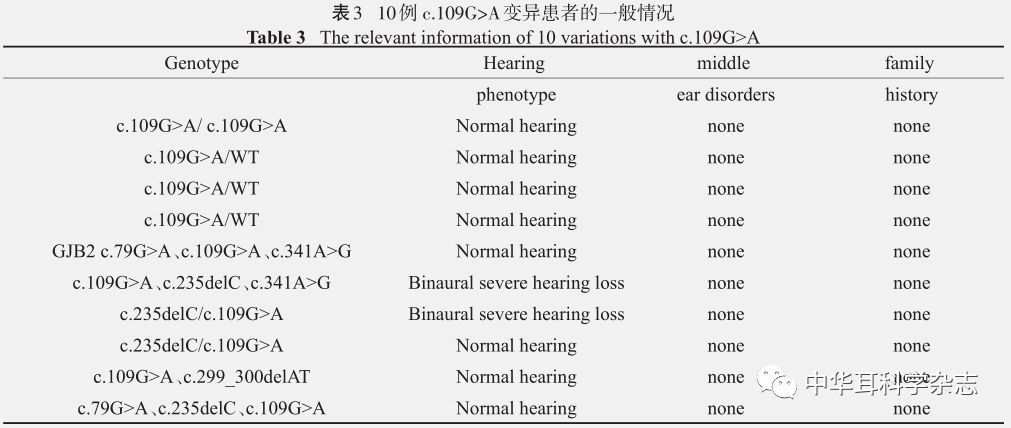
2.3 GJB2 c.109G>A基因型、听力表型等情况 本次共检测出10例携带c.109G>A变异,其中1例纯合变异,3例c.109G>A单杂合变异,6例复合杂合变异,具体情况如下表(表3)。
3讨 论 众所周知,基因变异具有普遍性、随机性、低频性、不定向性、多害少利性等特点,查阅文献及全国大样本数据显示我国最多见的遗传性耳聋相关变异基因为GJB2、SLC26A4、MT-RNR1,占耳聋相关基因的30-40%,成为遗传性耳聋的常见病因。本课题组前期研究中,取珠三角地区数个听障机构聋儿的样本行 9 项耳聋基因筛查,发现 GJB2与SLC26A4基因携带率极为接近[3],虽9项耳聋基因芯片覆盖了约80%的热点致聋基因及位点,但仍可能出现漏诊现象,因此,近期我们利用15项耳聋基因芯片技术联合一代测序技术对广州市某2个听障机构的家庭进行检测,旨在提高检测率,了解该家庭的可能分子构成,同时为患病家庭提供遗传咨询及生育指导。 通过本次芯片筛查,此机构最常见的致聋变异为GJB2,其次是SLC26A4,本次未检测出MT-RNR1及GJB3。其中GJB2 c.235delC(占总人数的14.11%),SLC26A4 c.919-2A>G(占总人数的6.64%)热点变异居多,与国内主流报道相似。由于GJB2突变存在明显的种族差异性,约49%的白种人家族常染色体隐性的耳聋是由GJB2导致的[4,5];高加索人热点突变为35delG,突变携带率为1.3%-2.8% [6];犹太人热点突变为167delT,在非综合征型耳聋中的突变率占据53% [7]。通过金标准的Sanger测序验证,除芯片包含的最常见位点外,还检测出了c.109G>A变异。目前,对c.109G>A的研究存在争议,大部分研究者趋向将 c.109G>A 错义突变归于致病变异。他们认为c.109G>A纯合突变的耳聋患者,表达产物缝隙连接蛋白的粘连受到影响,无法形成缝隙连接通道而导致耳聋[8]。携带c.109G>A变异的听力表型有异质性,本次共检测出 10 例携带c.109G>A变异,其中1例纯合变异为一位聋儿家长,双耳听力正常,中耳未见明显病变,无家族遗传史。另外3例携带c.109G>A单杂合变异的聋儿家长,亦无听力下降;仅有1例c.109G>A、c.235delC、c.341A>G复合与1例c.109G>A/c.235delC先证者为双耳重度聋患者。国内统计正常人群中携带c.109G>A的比例为12.5%,本次检测有10例,说明携带的比率较大,对耳聋基因的筛查有一定的参考意义,可能存在某些修饰基因[9]或其他调控区变异共同作用导致表型的异质性。另外,一般认为双等位基因突变可以导致表型的出现,对于大多数GJB2 c.235delC纯合突变的患者表现为语前重度-极重度聋,仅极少表现为语后中度聋,但在一个Trios家系中,听力正常、c.235delC单杂合的父母育有一对子女均为c.235delC纯合突变(芯片+Sanger测序),先证者7岁的妹妹却双耳听力均正常,实属罕见。国内解放军总院有报道 [10]1例38岁左耳突聋的c.235delC纯合突变患者听力表型为迟发型中度聋,并分析表型差异的可能机制为Connexin30过表达的补偿作用及修饰基因作用。因此,为探索其机制,本病例仍需继续进一步研究。 SLC26A4基因由于有21个外显子,变异存在广泛的等位基因异质性,中国人最常见的c.919-2A>G及c.2168A>G突变位点主要集中在7、8、19外显子上,白人中c.1246A>C、p.L236P和lVS8+1G>A是Pendred 综合征患者最常见的几种突变类型[11],日本c.2168A>G [12]和蒙古p.L676Q [13]是突变频率较高的类型。本次c.919-2A>G的检出率在SLC26A4基因中最高,成为选取的听障机构中第二主要病因,再次验证与我国的SLC26A4基因突变频谱[14]一致。十五项耳聋基因芯片较原来的9个位点筛选项目多出了6个SLC26A4高频位点,因此相对以往该基因的检出率提高,本次检测出有c.1229C>T变异,Sanger测序结果还发现c.259G>T、c.754T>C的已知致病突变。本研究中基因芯片筛查的SLC26A4单杂合先证者经过Sanger测序,结果发现3例携带双等位基因致聋,CT验证为双侧前庭水管扩大伴Mondini畸形;仍有先证者为单杂合致聋,对于这部分单杂合突变引起表型的改变,可能在启动子区或隐藏的剪接位点上存在某一些未被检测到的突变位点;或者其他未知基因共同发挥作用;或者与单核苷酸多态性、拷贝数变异有关;还有可能由于环境因素的影响[15]。因此需要进行进一步研究。 虽然本次未检测出MT-RNR1及GJB3基因,仅能说明本研究选取的听障机构内暂无MT-RNR1及GJB3基因的常见变异,间接证明MT-RNR1及GJB3基因致聋的发生率远低于 GJB2与 SLC26A4基因,并且随着医疗技术的发展及对医学常识的普及,在广州这个经济相对发达的城市,氨基糖甙类药物的使用明显减少,因此发生药物性耳聋的几率也相对下降;而GJB3基因变异的致病性向来存在质疑,有新研究报道,c.538C>T不一定和非综合征型遗传性耳聋相关[16]。从本次结果看,Sanger测序提高了致聋基因的检出率,芯片筛查结合金标准检测我们为部分家庭明确了病因,同时按照孟德尔遗传规律,为有再生育的家庭提供了遗传咨询。对于明确先证者为GJB2与SLC26A4基因致聋病因的家庭,有再生育计划时,可实施产前诊断、胚胎植入前基因诊断(PGD)或无创产前检测(NIPT)[17];而此类先证者,在选择结婚对象时,双方均应该进行耳聋相关基因检测,建议尽量避免同基因型双方进行婚配;并且在孕前、育前、怀孕期间、分娩后的新生儿,均应进行准确的遗传咨询,避免生育患者同样表型的患者。 本研究通过十五项耳聋基因芯片联合一代测序技术对广州市两所听障机构的Trios家系进行常见遗传性耳聋热点致病基因筛查并验证,结果提示广州市此特殊听障机构的耳聋相关基因热点变异仍以GJB2 c.235delC与SLC26A4 c.919-2A>G最常见,其他非热点变异也占一定比例。在大规模高危人群筛查、新生儿及孕妇筛查中,使用十五项耳聋基因芯片能快速、准确的检测出热点变异;由于基因芯片的检测位点有限,可联合一代测序提高非综合征型耳聋的变异检出率并验证准确性;对常见耳聋基因检测阴性的个体,可采取目标基因外显子捕获及测序(NGS panel)进一步帮助患者明确病因[18];有条件的家庭或实验室还可对NGS panel阴性的个体进行全外显子或全基因组,并进行拷贝数变异(CNV)检测分析。总而言之,在这个信息高速发展的时代,检测技术的成本下降,医务工作者应联合多种技术,中和利弊,为患者提供准确可靠的耳聋基因诊断、开展专业的遗传咨询、婚育指导、耳聋风险预测、干预出生缺陷。返回搜狐,查看更多 |
【本文地址】
今日新闻 |
推荐新闻 |