scRNA |
您所在的位置:网站首页 › 转录组学分析原理 › scRNA |
scRNA
|
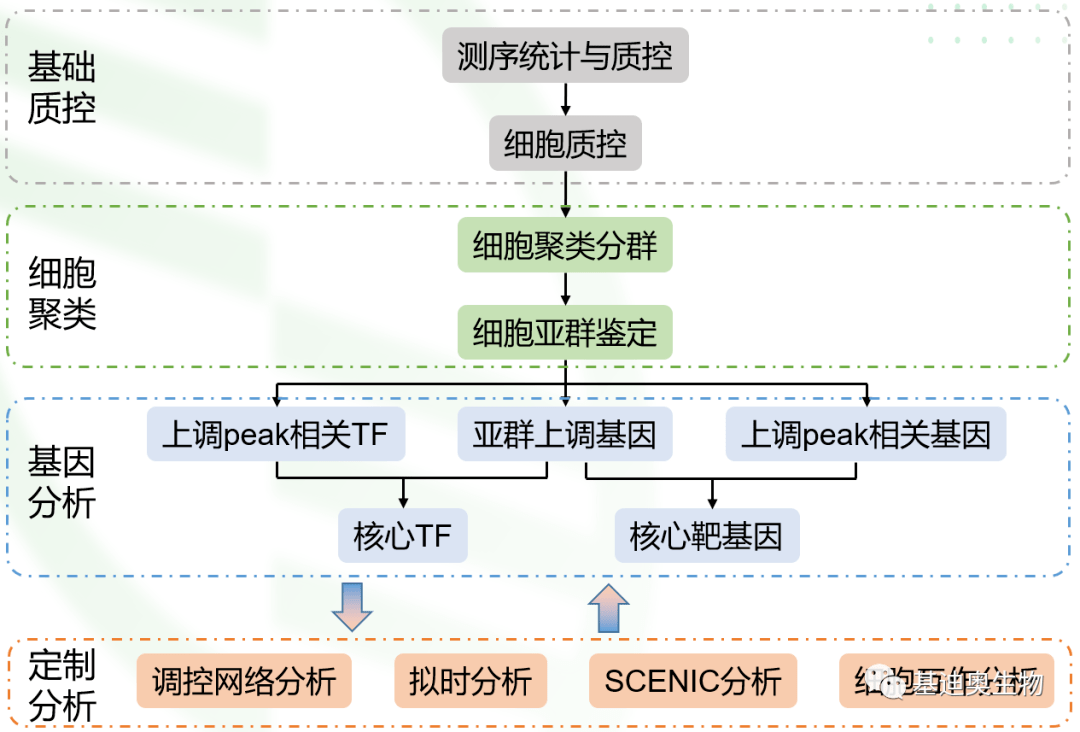
图2 关联分析的逻辑 2. 关联分析思路: 主要从基因上游的开放染色质区域容易结合转录因子来调控基因表达为核心主线的分析思路。其中关键步骤是通过两组学进行核心TF和核心靶基因的筛选,然后构建TF-Target gene调控网络。最后也可以将调控网络分析与拟时分析、细胞互作分析进行关联,从多分析点研究调控网络。
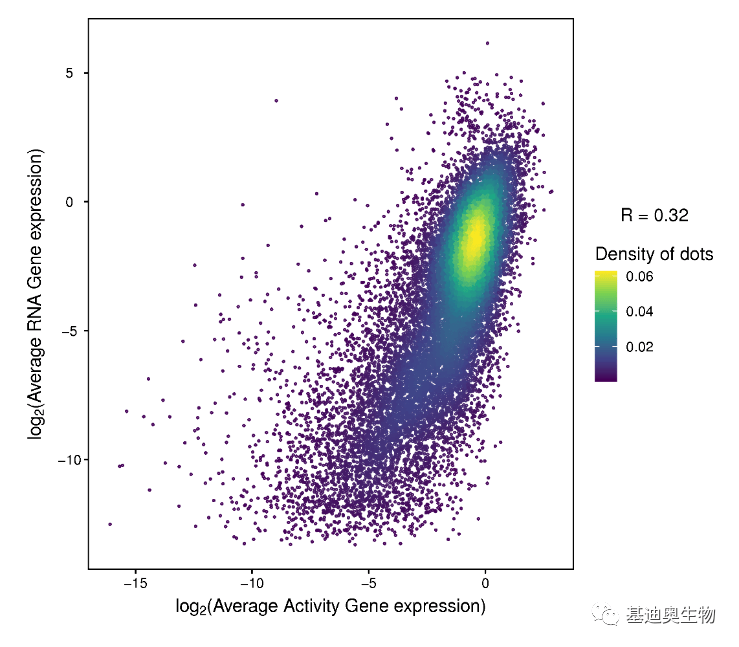
图3 单细胞RNA+ATAC关联分析思路 3. 思路应用 根据上面所述的两组学关联的数据分析思路,我们就可以从以下4个方面探索生物学问题: (1)更深入地鉴定细胞类型和状态。通过表观基因组和基因表达谱的综合分析来鉴定复杂的细胞群体,并捕获细胞异质性,从而发现隐藏的信息。基因表达标志物可帮助我们更轻松地解释表观遗传图谱。 (2)发现新的基因调控相互作用。将调控元件的发现与基因表达相结合,探索驱动细胞分化、发育和疾病的基因调控相互作用。 (3)探索细胞分化的驱动基因。了解罕见细胞中的基因调控网络及其分化调控。 (4)充分挖掘样本中的信息。多组学获得转录组和开放染色质信息,让我们从多组学中发现单组学意想不到的信息。 二、关键分析内容详情 1. 样本两组学相关性分析 在scATAC-seq数据中,对于每一个细胞,我们通过基因及基因上游2kb内的peak丰度去量化每个基因在每个细胞中基因开放性,即在这个区域内,peak丰度越高,基因就越有可能受到转录因子调控或与RNA聚合酶结合,基因开放性越高。对于每一个样本,我们计算平均基因开放性(scATAC-seq)和平均基因表达量(scRNA-seq),并进行相关性分析,使用pearson相关性系数来衡量每个样本两组学的相关性强弱。
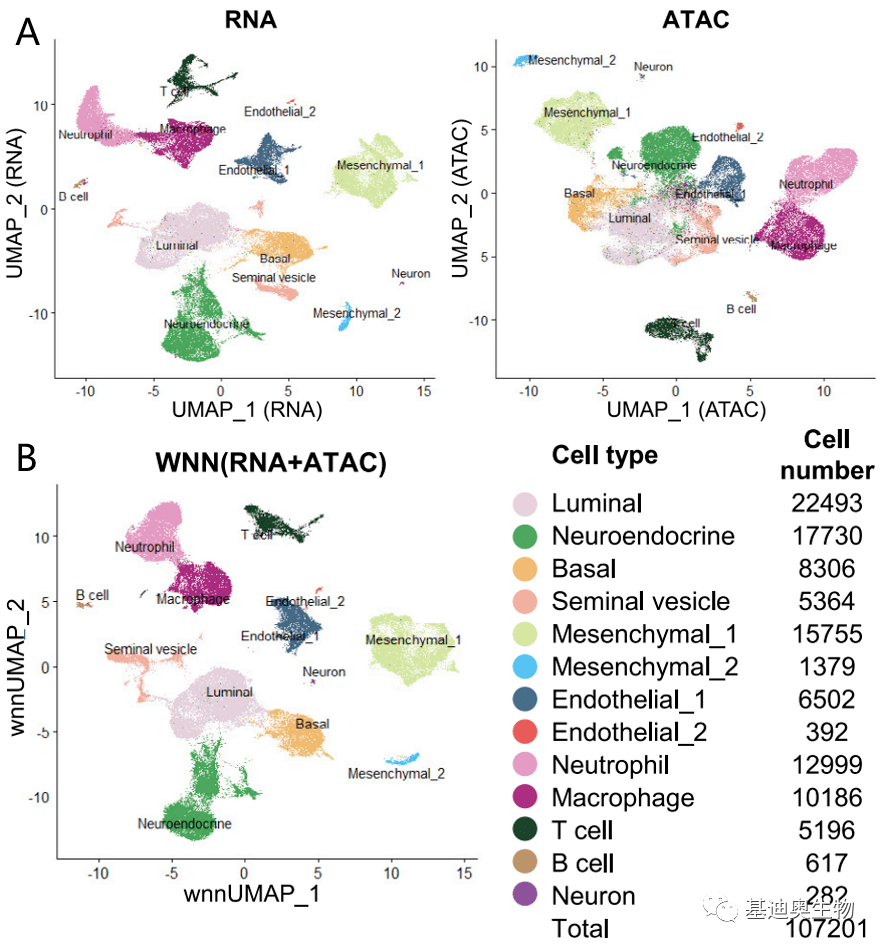
图4 两组学基因开放性和表达量的相关性散点图 2. 两组学整合的细胞图谱 一般来说,我们先对scRNA-seq数据聚类和注释,然后通过scRNA-seq注释结果再对scATAC-seq数据进行注释,最后我们将scRNA-seq+scATAC-seq数据整合,得到单个细胞中的基因表达和染色质开放性数据,通过WNN算法将相似的细胞聚类到一起,形成两组学整合的细胞图谱。从整合的细胞图谱中我们可以研究两组学的一致性,发现两组学一致的细胞类型特异性标记基因,也可能从ATAC数据发现RNA没有发现的亚群。
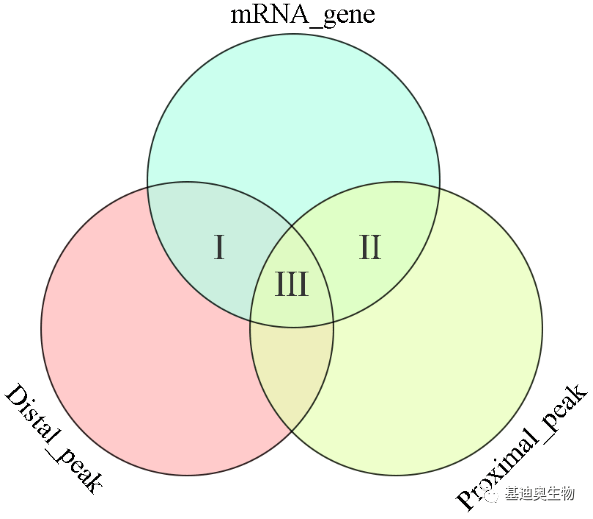
图5 整合的细胞图谱 3. 候选靶基因筛选 我们将在scRNA-seq数据中基因表达量上调且在scATAC-seq数据中基因近端(基因区及基因上游2kb)和远端(基因上游2-100kb)染色质可及性发生变化的基因定义为候选靶基因。这些基因的相关染色质可及性发生变化,同时表达量也发生,是潜在的受到染色质可及性调控的基因。这些基因又可以进一步划分为三个类别: (1)远端受到调控的基因,候选靶基因表达量上调,且基因远端具有上调peak,即维恩图中Ⅰ区域。这部分基因可能受到远端染色质开放性变化的影响,如远端增强子序列开放增强转录强度。 (2)近端受到调控的基因,候选靶基因表达量上调,且基因近端具有上调peak,即维恩图中Ⅱ区域。这部分基因可能受到近端染色质开放性变化的影响,如启动子序列开放启动转录程序、增强子序列开放增强转录强度。 (3)近端远端同时受到调控的基因,候选靶基因表达量上调,且基因远端和近端同时具有上调peak,即维恩图中Ⅲ区域。这部分基因可能同时受到近端和远端染色质开放性变化的影响,如TF通过远端染色质调节核小体重塑并引起基因近端染色质开放。
图6 候选靶基因示意图 4. 候选转录因子筛选 我们将细胞亚群中表达量上调,且对应motif在上调peak中富集的转录因子定义为候选转录因子。这部分转录因子的表达量水平上升,意味着转录调控机制的启动;富集相应motif的peak上调为这些转录因子的发挥作用提供了结合位点;所以,这部分转录因子具有参与转录调控的可能性。根据富集转录因子motif的peak所注释的靶基因位置,我们可以将候选转录因子进一步分为对靶基因进行近端调控的转录因子和对靶基因进行远端调控的转录因子(该划分方法与TF和靶基因的相对位置有关,并不是TF的固有属性。例,ATF4对基因ABCA6是近端调控TF,但对ABTB2是远端调控TF。) 5. 调控网络构建 (1)我们可以通过peak将候选靶基因和候选转录因子联系起来,从中筛选出核心基因。核心基因分为两类——核心转录因子和核心靶基因,核心转录因子可以结合到染色质开放区域(即peak),同时,这一个染色质开放区域又具有调控下游基因(核心靶基因)的可能,这样的TF-靶基因关系就构成了核心基因的调控网络。 (2)接着也可以进一步通过UMAP图来可视化核心基因及关联peak的分布情况,通过分布情况进一步确认核心基因之间的关系。若转录因子、peak、靶基因的分布情况一致,则说明筛选得到的核心基因有很大的可能存在调控关系,更容易在分子实验中得到验证。 (3)最后根据筛选得到的核心基因之间的调控关系,我们可以使用网络图来对该调控关系进行可视化。
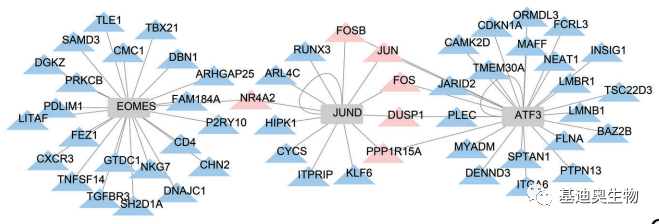
图7 TF与靶基因的调控网络图 6. 与其他分析的关联 (1)拟时分析:我们将亚群构建的regulons在AUCell中进行打分,然后将每个细胞与拟时间轴的打分进行非线性拟合,从而研究regulons在拟时间轴上的表达模式,挑选具有变化趋势较大的regulons在拟时间上进行发育表达调控的研究。
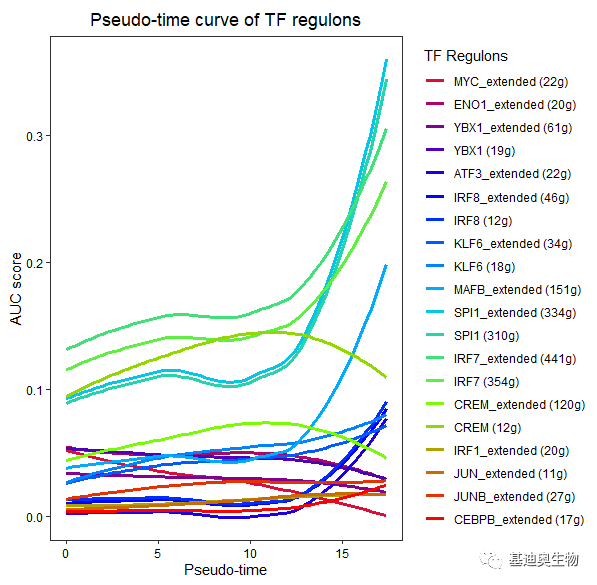
图8 regulons在拟时间轴上的非线性曲线 (2)拟时分析:不同细胞类型的分化是由发育过程中不同时间段的regulons塑造的,这些regulons以一个“主”TF为中心,通过DNA结合和转录诱导激活其细胞分化。因此,我们测试每个分支节点的差异regulons的重要性来破译regulons的树状图中的复合regulons,检验方法是Wilcoxon检验和Logreg检验,最终推导出regulons的动态表达程序,同时也可以与拟时分化轨迹的亚群进行一致性比较,从而可以筛选出最高表达下的祖细胞的主要细胞系,也可以用下游细胞亚群中特异的regulons定义细胞亚群名称,此外也可以研究拟时分化过程中的调控网络。
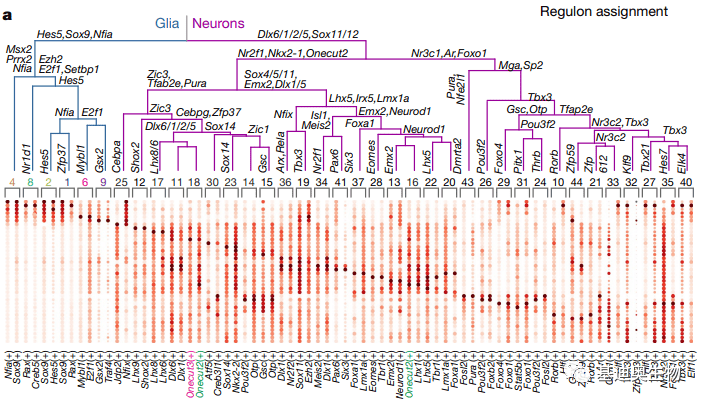
图9 regulons分配树状图 (3)SCENIC分析:我们可以与SCENIC分析的regulons找重合TF及靶基因,也算是两个分析结果的相互验证,也可以更进一步的缩小调控网络的研究范围。 (4)细胞互作:我们可以先通过cellphoneDB研究细胞间的配受体关系来找到有互作关系的细胞对,然后结合下游软件CellSign,用以分析受体潜在影响的转录因子,这为胞外信号和胞内信号的联系提供了方法,这可能更有利于对细胞通讯的深入理解。 单细胞测序和关联分析都可以联系基迪奥客户~ *未经许可,不得以任何方式复制或抄袭本篇文章之部分或全部内容。版权所有,侵权必究。 基迪奥生物|专业定制测序服务 联系方式:020-39341079;[email protected]返回搜狐,查看更多 |
【本文地址】