LeafGo: Leaf to Genome,一个快速的工作流程,使用长读测序技术生产高质量的从头植物基因组 |
您所在的位置:网站首页 › clr测序 › LeafGo: Leaf to Genome,一个快速的工作流程,使用长读测序技术生产高质量的从头植物基因组 |
LeafGo: Leaf to Genome,一个快速的工作流程,使用长读测序技术生产高质量的从头植物基因组
|
长读测序最具挑战性的方面之一是在实验室工作流程的每一步中施加严格的质量控制,以获得良好质量的测序结果。我们概述了提取HMW DNA的最佳条件,并将其处理成PACBIO和ONT平台的长读测序库。特别是,我们制备了PACBIO连续读(CLR)和高保真(HIFI,也称为循环共识序列,CCS)文库,并在最新的PACBIO平台,续集I和续集II上测序它们。为了GydF4y2Ba桉树GydF4y2Ba利用GridION平台制作和测序文库,证明了LeafGo实验室组分对ONT测序的适用性[GydF4y2Ba34GydF4y2Ba那GydF4y2Ba37GydF4y2Ba那GydF4y2Ba38GydF4y2Ba](见附加文件GydF4y2Ba1GydF4y2Ba:牛津纳米孔技术测序;表S2和S3;图S1)。最后,我们使用最新的基因组装配工具进行比较CLR和HIFI数据并组装了这两个GydF4y2Ba桉树GydF4y2Ba物种和GydF4y2Ba答:hypogaeaGydF4y2Ba进入高质量的基因组草案。GydF4y2Ba DNA提取,质量控制,图书馆和长读测序GydF4y2Ba DNA提取GydF4y2Ba叶片中实施的提取方案在一天内产生大量的HMW DNA,并使用最小的资源和努力。该方案在不同技术人员在不同的日子中产生了27种不同植物物种的27种单独的HMW DNA。产量(每1g叶片湿重)的范围为10至278μg(平均值±标准偏差[SD],79.6±71.6),具有高变异性GydF4y2Ba桉树GydF4y2Ba种,与其他种比较(图。GydF4y2Ba2GydF4y2Ba一种)。所有提取的HMW DNA都具有高纯度和完整性,尽管不同的组合物,包括潜在的污染物,物种之间[GydF4y2Ba12GydF4y2Ba那GydF4y2Ba29GydF4y2Ba那GydF4y2Ba32GydF4y2Ba那GydF4y2Ba34GydF4y2Ba那GydF4y2Ba36GydF4y2Ba].值得注意的是,这是GydF4y2Ba桉树GydF4y2Ba样品在裂解过程中显示出氧化的迹象,如溶液颜色变暗;然而,在不需要添加抗氧化剂的情况下,DNA的质量并没有受到影响[GydF4y2Ba12GydF4y2Ba那GydF4y2Ba34GydF4y2Ba].吸光度比表明污染物水平低,如蛋白质、碳水化合物和酚类,和高纯度(平均)GydF4y2Ba一种GydF4y2Ba260/280GydF4y2Ba= 1.83±0.05 SDGydF4y2Ba一种GydF4y2Ba260/230GydF4y2Ba= 2.21±0.13 sd;无花果。GydF4y2Ba2GydF4y2Bab)[GydF4y2Ba9.GydF4y2Ba那GydF4y2Ba34GydF4y2Ba那GydF4y2Ba36GydF4y2Ba那GydF4y2Ba39GydF4y2Ba].通过脉冲场凝胶电泳和毛细管电泳评估提取的基因组DNA(GDNA)的完整性。提取的样品通常显示出从大约> 15至1GydF4y2Ba:图。S2和S3)。我们的结果与DNA产率,片段长度和纯度术语方面的结果相当或改善,但具有更简单和更少的毒性提取方案[GydF4y2Ba12GydF4y2Ba那GydF4y2Ba17GydF4y2Ba那GydF4y2Ba34GydF4y2Ba],在1天内产生适合长读测序的HMW gDNA。GydF4y2Ba 图2GydF4y2Ba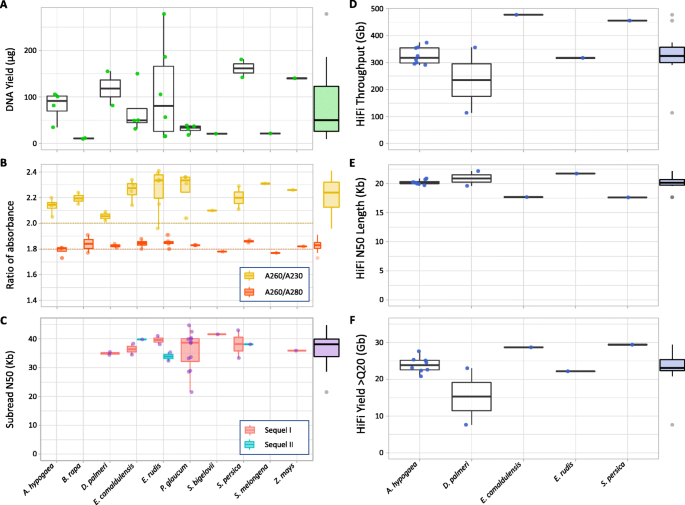
研究植物物种中的DNA提取和长读取测序输出。屈服 (GydF4y2Ba一种GydF4y2Ba)和吸光度比(GydF4y2BaB.GydF4y2Ba)来自十种研究植物物种的提取的HMW DNA。CLR文库的亚次N50和续集I和续集II的10个研究植物物种中的六种(GydF4y2BaCGydF4y2Ba).总吞吐量(GydF4y2BaD.GydF4y2Ba),子读N50长度(GydF4y2BaE.GydF4y2Ba)和Q20产量(GydF4y2BaFGydF4y2Ba)获取在Sequel II上测序的5种研究植物的HiFi文库。所有数据的平均值沿边缘标绘。CLR测序未完成GydF4y2Ba答:hypogaeaGydF4y2Ba那GydF4y2BaB. Rapa,GydF4y2Ba和GydF4y2Ba美国melongenaGydF4y2Ba 全尺寸图像GydF4y2Ba PacBio CLR和HiFi测序GydF4y2Ba来自八种物种的提取的HMW GDNA(GydF4y2BaArachis hypogaea,Distichlis palmeri,桉树鲁西斯,E.Camaldulensis,Pennisetum Glaucum,Salicornia Bigelovii,Salvadora Persica,GydF4y2Ba和GydF4y2BaZea Mays.GydF4y2Ba),处理成26个CLR和7个HiFi文库,然后用PacBio Sequel I (22: 1M SMRT细胞)和Sequel II (17: 8M SMRT细胞)进行测序。SMRTLink分析(附加文件GydF4y2Ba1GydF4y2Ba:表S4)显示CLR和HiFi文库的序列统计均高于PacBio推荐标准[GydF4y2Ba40GydF4y2Ba],并采用最优内控指标,表明测序反应未见抑制。GydF4y2Ba CLR库(附加文件GydF4y2Ba1GydF4y2Ba:图。S4a至S4d)显示了一种模式> 30kb,片段很少。测序结果证实,提取的GDNA具有良好的纯度和高分子量。每个续集I SMRT电池的平均CLR产量为11.5 GB(±4.7GB [SD]; 3.3-18.1 GB [MIN-MAX]),而续集II产生167.6GB / SMRT细胞(±26.1 GB; 140.8-195.4 GB)(附加文件GydF4y2Ba1GydF4y2Ba:图S4E和S4F)。平均N50子读长度为36.6 Kb(±5.5 Kb;21.5-44.7 Kb)为22个Sequel I CLR库,36.4 Kb(±3.2 Kb;32.4-39.8 Kb)的四个Sequel II CLR库(图。GydF4y2Ba2GydF4y2BaC)。测试CLR文库制剂的剪切DNA;然而,由于ont库以前所示,产量或n50没有明显的改善[GydF4y2Ba34GydF4y2Ba],而未剪切的DNA则用于所有其他文库。为了测试库加载是否影响子读N50长度,一些SMRT单元故意欠载。有一个边缘显著相关(附加文件GydF4y2Ba1GydF4y2Ba:表S5和图3. S5)ZMW(零模式波导)占用或库欠载(高p0%)和副N50(Spearman的Rho = 0.42GydF4y2BaP.GydF4y2Ba= 0.0499),表明以吞吐量产量为代价,子读N50的收益很小;不出所料,库加载(由P0%和P1%确定)与吞吐量产量显著相关(斯皮尔曼的rho =−0.84和0.86,GydF4y2BaP.GydF4y2Ba-Value HiFi库显示大约20 Kb的模式(附加文件GydF4y2Ba1GydF4y2Ba:图S6),当用Sequel II测序时,平均总吞吐量为322.9 Gb/SMRT细胞(±87.6 Gb;113.9-477.3 Gb), Q20产率23.3 Gb(±5.4 Gb;7.6-29.4 Gb)和子读N50的20.1 Kb(±1.3 Kb;17.6-22.2 Kb)如图所示。GydF4y2Ba2GydF4y2BaD-F。除了一个外,所有样品的总吞吐量和Q20产量都很高GydF4y2Bad . palmeriGydF4y2BaHiFi库负载不足(P1% = 13%);随后对装载最优的库进行重测序,得到了更典型的结果(356.3 Gb总吞吐量;23 Gb Q20产量)。GydF4y2Ba 从头基因组组装GydF4y2Ba桉树GydF4y2Ba和GydF4y2Ba落花生GydF4y2Ba物种GydF4y2Ba组装一个全新的基因组需要覆盖范围、读取长度、基础质量和计算资源的组合。准确的重建,最好是在短时间内,确实是至关重要的,因为组装的连续性和碱基的准确性都会影响基因组的质量。LeafGo的生物信息学建议是基于对现有和最近开发的工具的测试,以优化基因组质量和计算时间,这是一种通常有限的资源。为了评估最佳方法,评估并组装了两个二倍体桉树(2n = 2x = 20)和异源四倍体花生(2n = 4x = 40)的HiFi和CLR测序数据。GydF4y2Ba 原始测序数据的质量控制GydF4y2Ba第一生物信息学阶段是原始测序数据的质量控制(QC)。特定于平台的指标代表了排序运行的整体质量的第一个信息统计信息(附加文件GydF4y2Ba1GydF4y2Ba:表S4)。然而,使用LongQC执行了更全面的QC步骤[GydF4y2Ba41.GydF4y2Ba].LongQC或类似的软件,允许对原始测序数据进行快速和深入的跨平台QC。LongQC结果为GydF4y2Ba桉树GydF4y2Ba物种和GydF4y2Ba答:hypogaeaGydF4y2Ba排序数据显示在附加文件GydF4y2Ba1GydF4y2Ba:图S7和S8。GydF4y2Ba 三个物种的所有HiFi数据都是良好的质量,这表明了每读基调用准确性的高分(附加文件GydF4y2Ba1GydF4y2Ba:图S7A, S7F和S7M),除图中有轻微的双峰外,每个读覆盖率均为正态分布GydF4y2Bae·鲁迪GydF4y2Ba(附加文件GydF4y2Ba1GydF4y2Ba:图。S7C,S7H和S7O)。两个都GydF4y2Ba桉树GydF4y2Ba物种显示出类似的GC含量和序列复杂度(附加文件GydF4y2Ba1GydF4y2Ba:图S7B、S7G和S7D、S7I)。的GC内容GydF4y2Ba桉树GydF4y2Bareads在均值0.39(±0.03 [SD])附近呈明显的单峰分布,但在0.55附近有一个上子模态离群值。更仔细地检查这个较高的GC含量峰揭示了端粒重复的存在。GydF4y2Ba 的GC含量GydF4y2Ba落花生GydF4y2Ba(附加文件GydF4y2Ba1GydF4y2Ba:图。S7N)呈现尖锐的单峰分布,平均值为0.36(±0.05 [SD]),但观察到两个子模式:靠近0.55的上部模型异常,如桉树,一个下部模板异常近0.15。更接近较低子模式的研究显示了Centromere重复的存在[GydF4y2Ba42.GydF4y2Ba].任何数据集的侧面区域都没有人工序列适配器(附加文件GydF4y2Ba1GydF4y2Ba:图S7E、S7L和S7Q)。GydF4y2Ba 相比之下,两种桉树的CLR数据没有达到HiFi数据显示的相同质量水平。在CLR模式中不提供Phred评分,因此不能直接评估阅读基础呼叫的准确性。CLR的序列复杂性较低(在CLR和HiFi中,低复杂性序列分别为0-40%和0-20%),其侧边区域似乎扩展了超过100个碱基,表明可能存在人工序列(附加文件GydF4y2Ba1GydF4y2Ba:图S8B, S8F, S8D, S8H)。两个都GydF4y2Ba桉树GydF4y2Ba物种的CLR序列具有与HIFI数据相似的GC内容。所有读数的GC含量均具有尖锐的正态分布,左右的平均分布为40%(±4%),但在CLR分布中找不到指示端粒体重复并存在于HIFI数据中的上部模峰峰值。令人惊讶的是,CLR测序在检测比HIFI的体重复时可能不太敏感。GydF4y2Ba 基因组组装GydF4y2Ba在装配之前,GydF4y2BaK.GydF4y2Ba-mer计数可以洞察未知基因组的基因组大小。一种GydF4y2BaK.GydF4y2Ba-使用GenomeScope 2.0对HiFi数据进行mer评估[GydF4y2Ba36GydF4y2Ba的基因组大小GydF4y2Bae·鲁迪GydF4y2Ba那GydF4y2BaE. camaldulensis,GydF4y2Ba和GydF4y2Ba答:hypogaeaGydF4y2Ba分别为506 MB,510 MB和2.54 GB(附加文件GydF4y2Ba1GydF4y2Ba:图S9)。杂合度水平也用相同的方法估计GydF4y2BaK.GydF4y2Ba-mer方法,在两种桉树中均较高(ab:GydF4y2Ba大肠camaldulensisGydF4y2Ba, 2.19%;GydF4y2Bae·鲁迪GydF4y2Ba, 1.57%),预计将影响基因组组装[GydF4y2Ba37GydF4y2Ba]估计基因组大小[GydF4y2Ba38GydF4y2Ba].所显示的核苷酸杂合度模式GydF4y2Ba答:hypogaeaGydF4y2Ba遵循同种异体一体化基因组织的预期明显模式[GydF4y2Ba43.GydF4y2Ba)(附加文件GydF4y2Ba1GydF4y2Ba:图S9C)。GydF4y2Ba 这三个基因组是用针对长读序列优化的工具组装的:GydF4y2Ba44.GydF4y2Ba]对于CLR数据(只有两个桉树)和hifiasm [GydF4y2Ba45.GydF4y2Ba]对于HIFI数据。不同汇编程序之间的比较显示了CANU是CLR PACBIO数据的有效汇编器[GydF4y2Ba46.GydF4y2Ba].对于HiFi生成的数据,我们进行了不同汇编器之间的比较(hifiasm v0.8 [GydF4y2Ba45.GydF4y2Ba],hicanu v2 [GydF4y2Ba44.GydF4y2Ba, Flye v2.8.1 [GydF4y2Ba47.GydF4y2Ba, Wtdbg2 v2.5 [GydF4y2Ba48.GydF4y2Ba])发现Hifiasm优于其他汇编程序(附加文件GydF4y2Ba1GydF4y2Ba:表S6和S7)。GydF4y2Ba 桉树二倍体基因组GydF4y2Ba全基因组装配统计数据如表所示GydF4y2Ba1GydF4y2Ba.对于两种桉树,基于HIFI和CLR数据的组装基因组大小相似GydF4y2Bae·鲁迪GydF4y2Ba和GydF4y2Ba大肠camaldulensisGydF4y2Ba.HiFi组件优于CLR组件为两者GydF4y2Ba桉树GydF4y2Ba,它与其他物种基于hifi的组件达成了协议[GydF4y2Ba8.GydF4y2Ba那GydF4y2Ba44.GydF4y2Ba那GydF4y2Ba49.GydF4y2Ba].与CLR组件相比,N50/N90和L50/L90组件在HiFi组件中显示出明显更高的邻接性。此外,HiFi组件持续地产生最长的contigs (GydF4y2Bae·鲁迪GydF4y2Ba61.8 MB,33.7 MB;GydF4y2BaE. camaldulensis,GydF4y2Ba69.1 Mb、58.1 Mb;分别用于HiFi和CLR)。GydF4y2Ba 表1两个基因组装配统计GydF4y2Ba桉树GydF4y2Ba物种和GydF4y2Ba答:hypogaeaGydF4y2Ba.我们使用QUAST计算了装配统计信息。基于CLR的组件是3周期,详细介绍“GydF4y2Ba方法GydF4y2Ba“。结果基于吹扫组件(见“GydF4y2Ba方法GydF4y2Ba”)。GydF4y2Ba 全尺寸表GydF4y2Ba通过分离单倍型和清除单倍体估计组装单倍体基因组大小,并使用BUSCO评分评估完整性(附加文件)GydF4y2Ba1GydF4y2Ba:表S8)。基于初级包装的组装,单倍体基因组大小GydF4y2Bae·鲁迪GydF4y2Ba在518 MB(CLR)和549 MB(HIFI)之间变化,并产生1.77N的估计倍增性。对于替代品群(CLR,73.4%,HIFI,87.2%)表示,高完整的Busco分数(> 96%)和相对高的分数表示单倍型合理分离单倍型。另外,用于CLR组件的替代CONTIG集合显示了较低的完整性,进一步展示了HIFI组件的优越性。单倍体基因组大小GydF4y2Ba大肠camaldulensisGydF4y2Ba在CLR和HiFi主组件上分别为523和532 Mb。primary和alternative contig集均表现出较高的BUSCO评分(primary, > 97%;替代,> 93%)在CLR和HiFi组装。单倍清洗后装配的估计倍性超过1.98 N,结合BUSCO评分,表明该装配是全面的。对齐的GydF4y2Ba大肠茅GydF4y2Ba基因组(GydF4y2Ba33GydF4y2Ba显示我们的高保真GydF4y2Ba大肠camaldulensisGydF4y2Ba组装有9个完整染色体,其余两条染色体几乎完全跨越两个斑点(图。GydF4y2Ba3.GydF4y2Ba).类似地,GydF4y2Bae·鲁迪GydF4y2BaHIFI组件有五种全染色体,其余的染色体几乎完全跨越两到三个折叠。在计算时间方面,通过使用CLR进行同类覆盖的CLR组装与HIFI数据的组装基因组草稿(40×HIFI,53 CPU HRS; 50×CLR,3444 CPU HRS;GydF4y2Bae·鲁迪GydF4y2Ba).当CLR覆盖HiFi的时间增加4倍(51× HiFi, 81 CPU小时;230× CLR, 72491 CPU小时;GydF4y2Ba大肠camaldulensisGydF4y2Ba).这两个GydF4y2Ba桉树GydF4y2Ba基因组显示出相同的装配复杂度和相似的计算时间。这两个GydF4y2Ba桉树GydF4y2Ba进一步通过表型和GydF4y2Ba在网上GydF4y2Ba检查 [GydF4y2Ba50.GydF4y2Ba那GydF4y2Ba51.GydF4y2Ba](见附加文件GydF4y2Ba1GydF4y2Ba:表型和GydF4y2Ba在网上GydF4y2Ba鉴定GydF4y2Ba桉树GydF4y2Ba物种;图S10)。GydF4y2Ba 图3GydF4y2Ba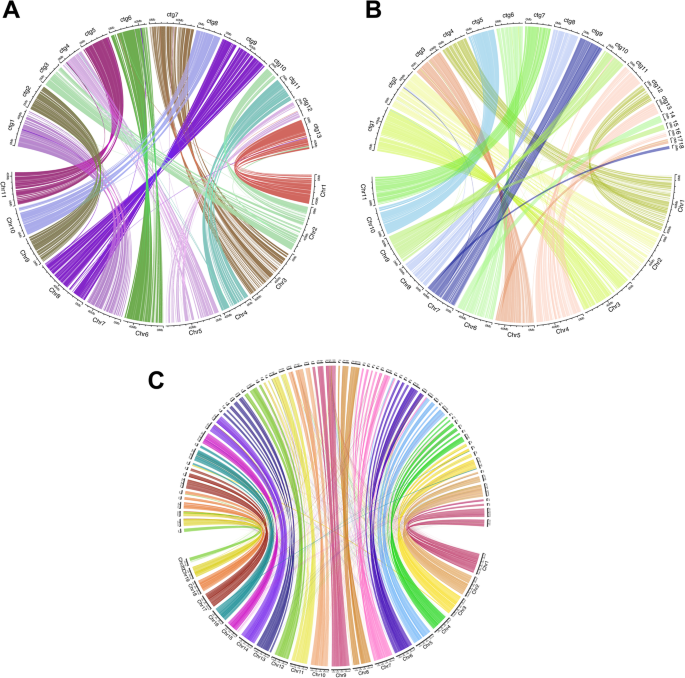
和弦图GydF4y2Ba大肠camaldulensiGydF4y2Ba年代,GydF4y2BaE. rudis和a. hypogaeaGydF4y2Ba根据参考基因组绘制的从头组装。对齐的GydF4y2Ba大肠camaldulensisGydF4y2Ba(GydF4y2Ba一种GydF4y2Ba),GydF4y2Bae·鲁迪GydF4y2Ba(GydF4y2BaB.GydF4y2Ba), 和GydF4y2Ba答:hypogaeaGydF4y2Ba(GydF4y2BaCGydF4y2Ba) HiFi组装反对GydF4y2Ba大肠茅GydF4y2Ba(GydF4y2Ba一种GydF4y2Ba和GydF4y2BaB.GydF4y2Ba),GydF4y2Ba答:hypogaeaGydF4y2Ba(GydF4y2BaCGydF4y2Ba)参考基因组GydF4y2Ba 全尺寸图像GydF4y2Ba最近的一会GydF4y2Ba大肠paucifloraGydF4y2Ba,类似规模和复杂性的基因组,可以与我们的组件进行比较。混合组装策略,illumina短读数产生的ont长读脚手架contigs,已经采用了生成的GydF4y2Ba大肠paucifloraGydF4y2Ba基因组(GydF4y2Ba36GydF4y2Ba].LeafGo方法更简单,劳动强度更低,只使用一种长读技术,产生的基因组的连续性是后者的12倍(contig N50 36/41 Mb vs 3.2 Mb,GydF4y2BaE.Rudis / Camaldulensis,E. PaucifloraGydF4y2Ba分别计算资源百分之一(见附加文件)GydF4y2Ba1GydF4y2Ba:基因组装配:计算资源)。GydF4y2Ba 花生异源四倍体基因组GydF4y2Ba同种异体一体化基因组的组装GydF4y2Ba答:hypogaeaGydF4y2Ba仅使用HIFI数据进行。这导致2623 MB基因组大小,分别具有42.3MB和10.37MB的CentIGN N50和N90。值得注意的是,这是GydF4y2Ba答:hypogaeaGydF4y2Ba基因组是一个具有丰富重复内容的大基因组[GydF4y2Ba25GydF4y2Ba]并且需要大量的长读取来帮助在基因组中弥合困难的区域。我们的大会证明比以前报告的大会仅由较旧的PACBIO技术产生更加连续,显示1.5 MB的N50,N90为0.34 MB [GydF4y2Ba25GydF4y2Ba].对齐的GydF4y2Ba答:hypogaeaGydF4y2Ba参考基因组(GCA_003086295.2)表明我们的HiFiGydF4y2Ba答:hypogaeaGydF4y2Ba组装是全面的,其中大多数染色体中的大部分都是跨越几个斑点的。对准的仔细检查显示了中央和外周染色体区域(可能的端粒和/或焦粒子),不完全组装成较大的凸起,因此缩短染色体水平组件(图。GydF4y2Ba3.GydF4y2BaC)。实际上,大会面临花生基因组重复性的挑战[GydF4y2Ba24GydF4y2Ba],富有重复含量而不是桉树(GydF4y2Bae·鲁迪GydF4y2Ba, 37.4%;GydF4y2Ba大肠camaldulensisGydF4y2Ba, 38.3%;GydF4y2Ba答:hypogaeaGydF4y2Ba,82.9%;额外的文件GydF4y2Ba1GydF4y2Ba:图S9)。虽然用HiFi数据组装大花生异源四倍体基因组的计算时间比较小的二倍体eucalyptus基因组更长(74× HiFi, 1081 CPU hrs,GydF4y2Ba答:hypogaeaGydF4y2Ba).GydF4y2Ba 时间估计GydF4y2Ba从RAW读取到HIFI / CCS的估计总时间读为0.6至1.0GB的二倍体基因组的高质量连续草案的组装,少于一天,其四重子基因组为2.5 GB的四倍体基因组增加到大约2天(查看其他文件GydF4y2Ba1GydF4y2Ba:基因组装配:计算资源)。当与HMW DNA提取的时间估计相结合(1天),HIFI文库制备和测序(5天)和组装,可以在最少7天内从植物样品中制备0.6-1.0 GB的高质量草案基因组。根据可用计算资源。该时间增加了多倍体(2.5 GB)基因组,其中测序需要大约9天,使时间长达16天(附加文件GydF4y2Ba1GydF4y2Ba:图S11)。所有时间估计都是基于使用一台Sequel II机器的测序,并根据覆盖率要求而有所不同。GydF4y2Ba |
【本文地址】