【临床试验百问系列】四十四 |
您所在的位置:网站首页 › 获得临床批件后多久正式批件完成检验 › 【临床试验百问系列】四十四 |
【临床试验百问系列】四十四
|
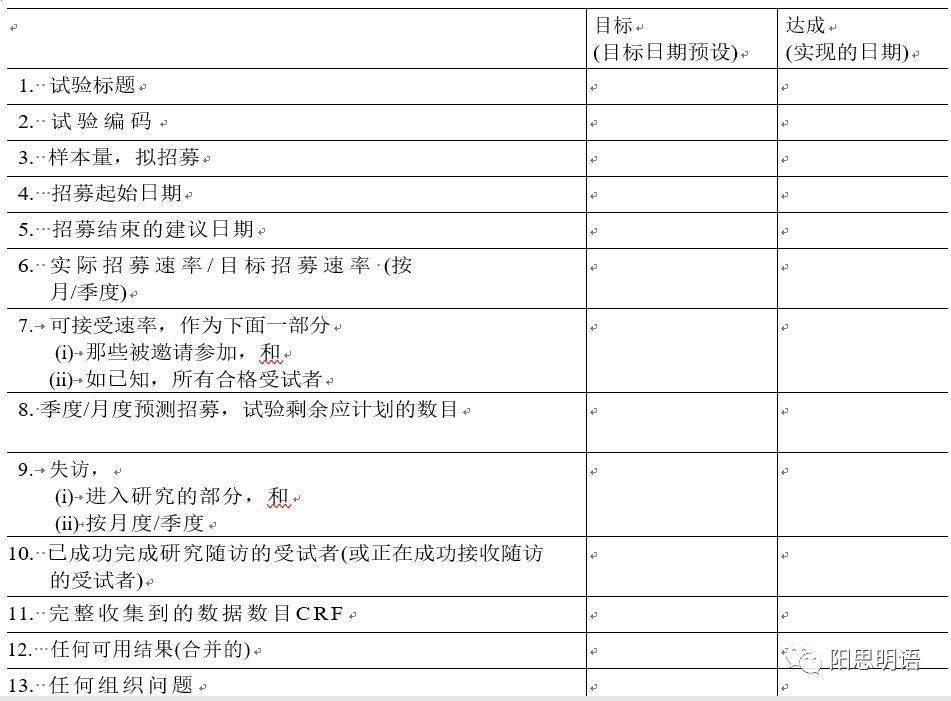
有兴趣的伦理委员会,可联系后台,获取该报告模板,丰富现有SOP。 伦理委员会应创建自己的表格或清单,要求提供研究者提供信息。如上所示,一般而言,伦理委员会要求的内容包括: -研究状态(例如,研究是否开放并且正在招募受试者?研究关闭了吗?正在进行数据分析吗?); -多少个筛选同意、入组、活跃、完成的受试者; -从研究中撤除的受试者数及其原因(尤其重视那些应无法耐受毒性和对研究缺乏疗效感到失望的情况,最容易被申办方忽略不提); -重大方案偏差/违背摘要以及采取的措施防止将来出现类似偏离/违背; -严重不良事件的描述和结果以及未曾预料到的涉及受试者或其他人的风险的问题; -确认当前方案(和批准的修正案)和同意书(适用的经批准的修订); -当前的风险效益评估; -自上次IRB审查以来,研究汇总的新信息(例如PD-1等同时开许多个试验,应当汇总相关信息,进行药物临床试验小结报告 ) 3 批件失效时的处理 按照ICH E6R2和FDA要求,在研究伦理批准到期之前,必须得到伦理审查并重新获得伦理批准研究。提前与伦理沟通获知开会日期和审评原则,是研究者和申办方/CRO的职责,也应当是伦理委员会公开、透明制度。中国已经加入ICH,应当在同一准则下行事。 如果申办方/CRO延误导致,或伦理委员会因不可预知原因延期开会,导致在伦理继续审查和批准之前,该伦理初始批件期满,则研究者不应入组新受试者参加到研究中来,直到获得伦理持续批准许可。这意味着,已完成入组的中心、处于随访期的中心,在这方面持续审查的频度和影响是较小的,可继续进行(发生重大非预期问题,研究者和申办方应当立即报告,这完全不冲突,是现实可行的)。 方案修订必须提交给EC进行审查和批准。修正案要求的变更不能立即实施,直到获得书面伦理批准文件(不能是口头通知,必须见到批件)。 对先前批准的同意书进行的更改也必须在使用修订后的文件之前,必须获得IEC批准。 该规则的例外是需要立即更改方案以消除对受试者安全和健康的明显直接危害。当方案更改基于此需求时,更改应立即实施,并必须随后通知IEC。 4 和《年度报告》一起需持续审核的信息 每家中心几乎只规定,报告PD、SAE和重度AE、年度报告。 在杜克大学、NCI、加州大学洛杉矶分校等组织,我们会发现一些较好做法,这些报告应当保存在ISF中,体现伦理动态、持续审查的沟通情况,既要看申办方、又要看研究者、还要看伦理委员会,因为FDA就是通过这三者来保护受试者的。 包括: •试验进度报告–研究者向伦理委员会提供的报告。研究者总结迄今为止的试验进展。这些报告频率由各自伦理确定,并且可能是根据研究涉及的风险程度。 •年度伦理更新–研究者提交的文件以获得继续进行研究的批准。规定要求伦理每个方案至少每年更新一次,一些伦理要求进行更频繁的审核。提交一个伦理申请递交函副本,继续研究批准记录需发送至申办方存档TMF。 •IND安全报告–由申办方生成并发送的报告。发生安全问题时,向所有中心研究者发送。IND安全报告副本应转发给伦理委员会;文件资料提交伦理委员会通知书应与IND安全报告副本一起保存在ISF。 欢迎机构、伦理、研究者展开合作,共同提升水平。我们正在翻译整理国际著名组织的《伦理审评手册》,欢迎加入到阳思明语CTBMI组织,共同完善。 我们以中国最专业的临床咨询服务,比国外咨询公司更低的价格,提供高质量服务,欢迎咨询合作。 提供全球视野的最优认知和最佳实践,可访问www.ctbmi.com(Web端打开才能看见)相关介绍。 CRA模块和CRC模块, Access至对应单模块文章,7200元,并包含十二场幻灯、CRA/CRC小册子。 PM、QA、MA模块,Access至 对应单模块文章,10000元,并包含十二场幻灯,相关小册子。 返回搜狐,查看更多 |

【本文地址】
今日新闻 |
推荐新闻 |