CDE:化学药品Ⅰ期临床试验申请药学研究信息汇总表(修订版)征求意见 点击上方的 行舟Drug ▲ 添加关注为了更好地实施国家局《关于调整药物临床试验审评审批程序的公告》(2018年第50号... |
您所在的位置:网站首页 › 药学CDE › CDE:化学药品Ⅰ期临床试验申请药学研究信息汇总表(修订版)征求意见 点击上方的 行舟Drug ▲ 添加关注为了更好地实施国家局《关于调整药物临床试验审评审批程序的公告》(2018年第50号... |
CDE:化学药品Ⅰ期临床试验申请药学研究信息汇总表(修订版)征求意见 点击上方的 行舟Drug ▲ 添加关注为了更好地实施国家局《关于调整药物临床试验审评审批程序的公告》(2018年第50号...
|
来源:雪球App,作者: 行舟Drug,(https://xueqiu.com/4396147139/150608809) 点击上方的 行舟Drug ▲ 添加关注
为了更好地实施国家局《关于调整药物临床试验审评审批程序的公告》(2018年第50号),促进创新药的研究和开发,对创新药I期临床试验申请中存在的与安全性内容相关的药学常见问题进行梳理,组织起草了《化学药品创新药I期临床试验申请药学共性问题相关技术要求》,并且对《新药I期临床试验申请技术指南》(2018年第16号)中所附《化学药品Ⅰ期临床试验申请药学研究信息汇总表》进行了修订,经中心内部研究并组织业界讨论,形成征求意见稿。 现向社会公开征求意见,征求意见时限为自发布之日起1个月。 请将您的反馈意见发到以下联系人的邮箱: 联系人:马春辉,姚方耀 联系方式:[email protected], [email protected] 感谢您的参与和大力支持。 国家药品监督管理局药品审评中心 2020年6月1日
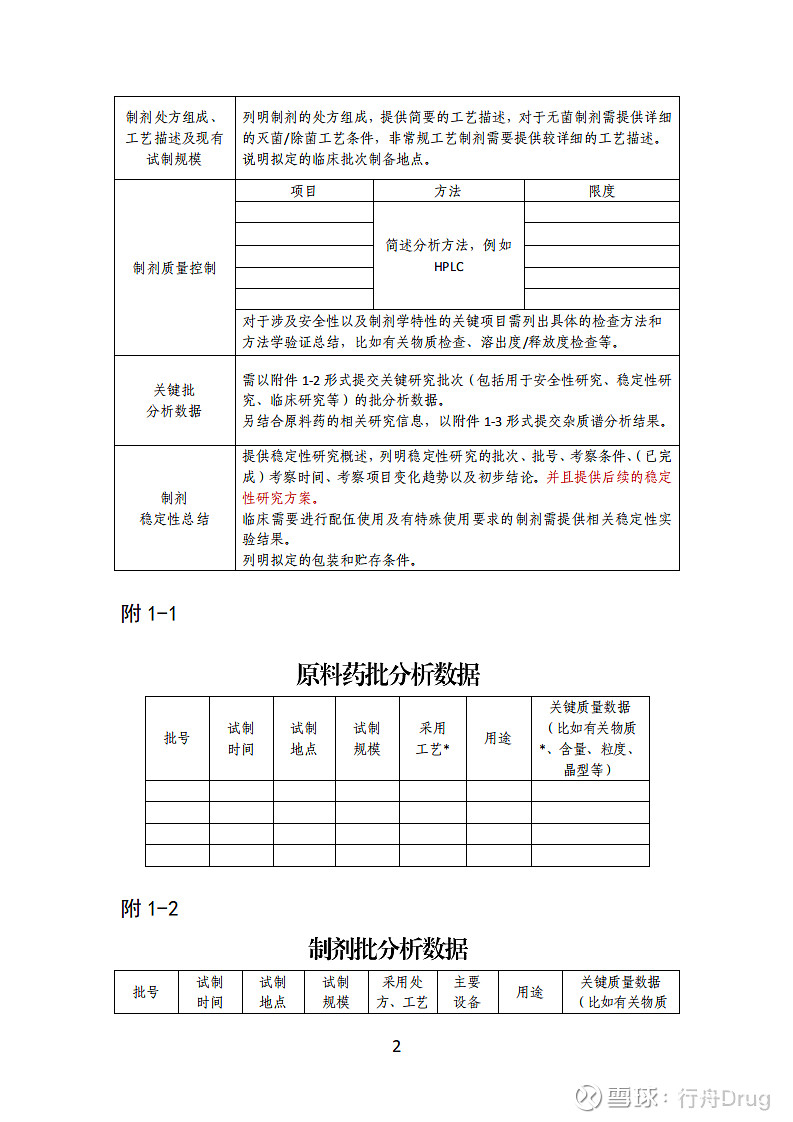
化学药品创新药 I 期临床试验申请药学共性问题相关技术要求(征求意见稿) 为鼓励创新,加快新药创制,满足公众用药需求,国家局发布了《关于调整药物临床试验审评审批程序的公告》(2018 年第 50 号),实行临床试验默许制以及 pre-IND 沟通交流制度。自该公告实施以来,符合要求的创新药 I 期临床试验申请均得到了快速审评(60 个工作日)并且顺利开展临床试验。 对于 I 期临床试验申请,为了保障受试者的安全,药学审评通常重点关注与安全性相关的问题,例如杂质、稳定性、无菌制剂生产条件和除菌/灭菌方法、以及临床前动物安全性评价试验与后续人体临床试验所用样品的质量可比性等。国家局发布的《新药 I 期临床试验申请技术指南》(2018 年第16 号)对相关药学研究内容和资料提交要求已经进行了阐述,但是审评中发现部分创新药 I 期临床试验申请仍然存在一些与上述安全性内容相关的药学问题。为了更好地实施国家局50 号文,促进创新药的研究和开发,本技术要求对创新药 I 期临床试验申请药学共性问题进行总结,以供申请人参考。 一、 关于样品试制 共性问题:提供的样品试制信息非常有限,处方工艺信息(特别是涉及复杂原料药或者复杂制剂时)过于简单。 一般性要求:参照《新药 I 期临床试验申请技术指南》相关要求提供原料药和制剂的生产商、生产地址和处方工艺信息,汇总关键研究批次(包括用于安全性研究、稳定性研究、临床研究(如已制备)等批次)的试制信息、关键质量数据等。 对于复杂原料药(例如多肽、小分子核酸、聚合物产品、含多个手性中心、含发酵工艺或者天然来源等药物)、复杂制 剂(例如微球/微乳/脂质体、胶束、透皮制剂、吸入制剂等)、复杂给药途径(例如制备成混悬液、乳液或者凝胶通过皮科、眼科和耳用等局部给药)以及复杂药械组合产品,应注意对重要的生产步骤、设备和工艺参数等进行较为详细的描述。对于无菌制剂,应对无菌生产条件和除菌/灭菌方法等进行较为详细的描述,并且提供无菌保障措施。 鉴于国内目前临床试验申请为 60 天默许制,I 期临床试验申请如果研究资料符合要求通常可快速开展临床试验,建议申报 I 期临床试验时(特别是涉及复杂原料药和制剂、复杂给药途径、药械组合产品时)在拟定的临床样品制备地点至少完成 1 批样品的制备,并且提供相关的试制信息、检验报告。如果拟定的临床样品制备地点与申报 I 期临床试验注册批次样品制备地点不同,注意对生产地址、设备、批量等不同可能影响产品质量桥接的风险进行评估,并且必要时在申请 pre-IND 沟通交流时提前与监管机构进行相关沟通。 二、 关于质量标准和分析方法 共性问题:仅简单提供质量标准,缺少提供部分关键项目的分析方法、未提供关键项目分析方法必要的验证信息。 一般性要求:参照《新药 I 期临床试验申请技术指南》相关要求研究并制定临床试验样品质量标准,以表格形式提供相关检测项目和可接受限度要求。注意提供关键项目(例如有关物质、残留溶剂、金属催化剂及金属试剂残留、遗传毒性杂质、溶出度/释放度、含量等)的具体分析方法,以及必要的方法学确认信息(例如至少包括专属性、灵敏度等)。 提供关键研究批次(包括用于安全性研究、稳定性研究、临床研究(如已制备)等批次)的检验报告。 三、关于杂质研究和控制 共性问题:未进行杂质谱分析或者分析过于简单,缺少对生产过程中用到的有毒有害试剂、金属催化剂等残留杂质的汇总分析。样品中的杂质记录不够详细,提供的检验信息中仅简单说明合格、未提供具体检测数据。未分析说明杂质限度拟定的初步依据。 一般性要求:参照《新药 I 期临床试验申请技术指南》 相关要求进行杂质研究并提供杂质谱分析信息。注意对产品中的工艺杂质、降解杂质、无机杂质/金属催化剂杂质、残留溶剂等进行汇总分析。同时,如果申报的原料药合成步骤较短,应注意加强对起始物料中可能引入杂质的研究和分析。 杂质记录方式可参照 ICH Q3A/Q3B,对于已鉴定结构的杂质按照单个已知杂质分别列出检验结果,未鉴定结构但固定出现的杂质按照相对保留时间列出检验结果,以便于样品之间杂质水平的桥接。提供的样品检验信息中应明确给出杂质的具体检测数据。 对于样品中实测值较高的杂质,应进行相关的安全性分析,并且在申报资料的药学及药理毒理部分中同时分别提交杂质限度拟定的安全性分析资料,包括安全限度的计算方式及确定依据。临床研究样品的杂质水平不得超出动物安全性试验数据所支持的相应的杂质水平。 四、关于遗传毒性杂质研究 共性问题:未进行遗传毒性杂质分析,或者对于潜在的遗传毒性杂质仅凭经验简单分析和界定为不具有遗传毒性。仅说明遗传毒性杂质含量符合要求,未提供具体的分析方法以及必要的方法学验证信息、检测数据等。 一般性要求:参照 ICH M7 指导原则对药品(包含起始物料制备)中的相关工艺杂质(例如起始物料、中间体、副产物)、降解产物和有毒有害试剂等的潜在遗传毒性进行初步分析和研究。建议参照 ICH M7 采用(Q)SAR 方法对结构已知的全部杂质进行预测和筛查。对于ICH M7 中 1 类、2 类杂质,应结合临床试验方案控制其不超过特定的可接受限度或者 TTC值。临床试验样品中的遗传毒性杂质水平需符合要求,保证临床试验阶段受试者的安全。 以表格的形式汇总提供潜在遗传毒性杂质研究分析信息、检测结果或者相关的控制策略。同时,提供遗传毒性杂质检测的分析方法以及必要的方法学验证资料。 五、关于稳定性试验 共性问题:提供的稳定性试验信息非常有限,不足以支持药品质量在计划的临床试验研究期间符合要求。对于注射剂以及一些具有特殊使用要求的制剂,未提供临床配伍稳定性的研究资料。 一般性要求:参照《新药 I 期临床试验申请技术指南》 相关要求开展药品稳定性试验并提供相关研究资料。应提供至少 1 批代表性批次(例如临床样品制备地点制备的样品、安全性研究批次)以及支持性研究批次(例如开发批次)的稳定性研究数据,同时提供后续的稳定性试验研究方案;对于不稳定的产品一般应尽可能提供多批次样品的稳定性研究数据。提供的稳定性研究数据以及后续的稳定性试验方案应能够支持药品质量在计划的临床试验研究期间符合要求。另外,需明确临床样品拟定的贮藏条件。 对于临床试验中需要进行配伍使用(例如注射剂)或者有特殊使用要求的制剂,一般应提供相关配伍稳定性试验或者使用中稳定性试验的初步研究结果,试验中注意关注配伍过程中新产生杂质的安全性以及注射剂不溶性微粒等情况。 六、关于安慰剂 共性问题:未说明临床试验中安慰剂的拟使用情况。 一般性要求:参照《新药 I 期临床试验申请技术指南》中相关要求,如临床试验方案中需使用安慰剂,应提供安慰剂的生产厂、处方工艺信息、质量控制等研究资料。同时,安慰剂需进行必要的稳定性试验考察。 七、关于辅料和包材 共性问题:使用到全新的辅料和包材,但未提供相关的资料。 一般性要求:参照《新药 I 期临床试验申请技术指南》中相关要求,I 期临床试验申请时,对于国内外制剂中尚未使用过的全新辅料,以及新材料、新结构、新用途的包材,需提供相关信息并按照相关文件要求进行关联申报。 上述问题为创新药 I 期临床试验申请中药学常见问题,建议申请人在研发和申报注册过程中予以关注。此外,根据国家局 50 号文,申报 I 期临床试验之前需进行 pre-IND 沟通交流,在此也建议申请人,如果新药研制过程中存在与上述安全性内容相关的问题或者疑问,可充分利用 pre-IND 沟通交流平台,提前与监管机构进行针对性的沟通交流。 参考文献: (1)《国家药品监督管理局关于调整药物临床试验审评审批程序的公告》(2018 年第 50 号); (2)《药物研发与技术审评沟通交流管理办法》(2018 年第74 号); (3)《新药 I 期临床试验申请技术指南》及《化学药品 I 期临床试验申请药学研究信息汇总表》(国家局 2018 年第 16号); (4)创新药药学研究的阶段性考虑,张宁,王亚敏,陈震, 《中国药学杂志》(2014 年 9 月第 49 卷第 17 期) ( 5 ) Guideline on the requirements for the chemical and pharmaceutical quality documentation concerning investigational medicinal products in clinical trials.(EMA) (6)Guidance for Industry Content and Format of Investigational New Drug Applications (INDs) for Phase 1 Studies of Drugs, Including Well-Characterized, Therapeutic, Biotechnology-derived Products.(FDA) (7)Formal Meetings Between FDA and ANDA Applicants of Complex Products Under GDUFA Guidance for Industry. (FDA) (8)ICH Q3A(R2)-Impurities in new drug substances. (9)ICH Q3B(R2)-Impurities in new drug Products. (10)ICH M7(R1)-Assessment and control of DNA reactive8 (mutagenic)impurities in pharmaceuticals to limit potential carcinogenic risk.
文章信息源于CDE,登载该文章目的为更广泛的传递行业信息,不代表赞同其观点或对其真实性负责。文章版权归原作者及原出处所有,文章内容仅供参考。本网拥有对此声明的最终解释权,若无意侵犯版权,请联系小编删除。 学如逆水行舟,不进则退; 心似平原走马,易放难收。 行舟Drug 每日更新 欢迎订阅+ 医药大数据|行业动态|政策解读 |



【本文地址】