【苏大检测】 |
您所在的位置:网站首页 › 美国fda申请受理要多久出结果呢 › 【苏大检测】 |
【苏大检测】
|
摘要:利用Q-Submission计划与FDA进行关于医疗器械申报前的沟通 合集:#FDA 2023年6月2日,美国FDA修订发布了Q-Submission Program指南文件(Request for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program)。该指南为医疗器械申办者提供了一种与FDA沟通的机制,通过该机制,申办者可要求FDA就医疗器械申请提供反馈或与之进行会议沟通。 Q-Submission,简称Q-Subs,主要有四种类型:Pre-Submissions(Pre-Subs)、Submission Issue Requests(SIRs)、Study Risk Determinations、Informational Meetings,分别简介如下。 A.Pre-Submission(Pre-Subs)
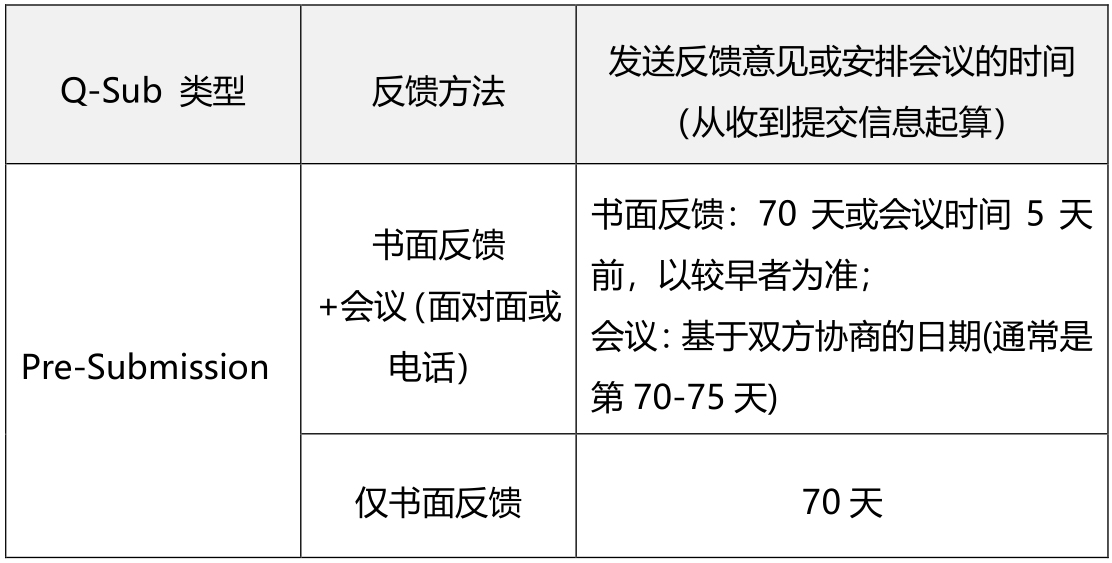
Pre-Subs适用的提交类型 Pre-Sub要求申办者提出正式书面申请,请求FDA给出书面反馈,或者申办者可以选择在书面反馈后进行会议,在会议纪要中记录任何额外的反馈或澄清。这种Pre-Sub会议可以是面对面的,也可以是电话会议。 Pre-Sub适用于申请提交前,包括上市申请(即IDE、PMA、HDE、De Novo、510(k)、Dual、BLA、IND)、附件分类请求或CW。
富有成效的Pre-Sub互动问题 促成有效反馈的提问通常具有如下特征: ● 要求对拟定方案进行具体的反馈; ● 已考虑到并包括引用适用的指南文件、标准和以前与FDA的讨论; ● 清楚地说明期望的结果,包括使用适应证或已标识用途; ● 在提交的材料中,为未来的器械开发和提交准备提供信息。 预计会促成富有成效的Pre-Sub互动的问题,通常为如下12类:法规监管战略问题、适应证/预期使用问题、临床研究问题、标识问题、再处理,灭菌&货架有效期问题、非临床台架性能测试问题、动物研究问题、生物相容性问题、软件/硬件问题、人因问题、网络问题。 提交者应注意:Pre-Subs中,FDA对特定问题的反馈将有助于指导产品开发和/或提交准备,但FDA并不准备对拟提交的材料进行预先审核或对拟提交材料中提供的数据进行预先审查。
B.Submission Issue Requests (SIRs)
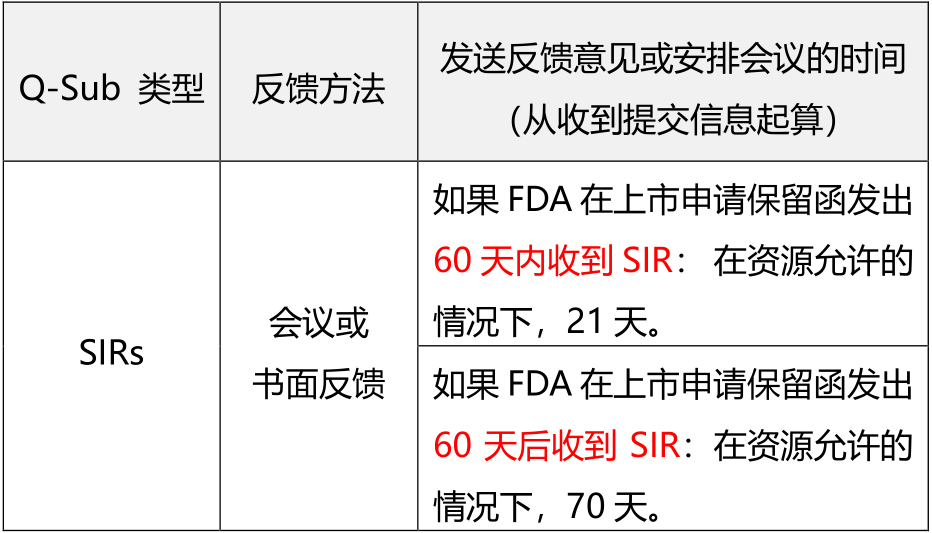
SIRs适用的提交类型 SIR是请求FDA对拟议的方法进行反馈,以解决上市申请保留函、CW保留函、IDE信函或IND临床保留函中传达的问题。前述上市申请即PMA、HDE、De Novo request、510(k)、Dual、BLA。 为了进一步阐明SIR的范围,在本指南中,以下内容被认为是适当的上市申请保留函情况: ● 510(k)s、De Novo请求、CWs和Duals所需的补充信息; ● 主要缺陷、不可批准、有缺陷可批准、可批准待定GMP,以及批准PAS条件下的PMAs和HDEs; ● 生物制剂许可申请(BLAs)的完整答复函。 SIRs注意事项 SIR旨在促进FDA和申办者之间的互动,以快速解决或澄清这些信函中确定的问题,以便项目能够向前推进,促使申报人能够在其正式答复中充分解决悬而未决的问题。但是SIR不应该被用来要求FDA预先审查拟提交的正式答复是否充分。无论是否提交SIR,提交者都应在要求的时间内对其收到的任何FDA信件做出正式回应。请注意,SIR不适合讨论传达最终决定的信件,如非实质性等同、撤回和删除。对于信件中不需要管理层参与的简单问题的澄清(例如,轻微的澄清问题或行政问题,可以由首席审查员解决),不需要SIR。当一个文件处于积极审查状态时,也不需要通过SIR来讨论问题。
C.Study Risk Determinations
Study Risk Determination是请求FDA确定一项计划中的医疗器械临床研究是否具有重大风险(SR)、非重大风险(NSR),或可根据IDE法规(21 CFR第812部分)予以豁免。对于不被豁免的研究,申办者负责做出最初的风险判断(SR或NSR),并将其提交给机构审查委员会(IRB)。FDA可帮助申办者、临床研究者和IRB进行风险确定。FDA是判定器械研究属于SR还是NSR的最终裁决者,其将在IDE申请提交后,或申办者、临床研究者或IRB询问时做出决定。
D.Informational Meeting
信息会议是期望与FDA进行信息分享,无需得到FDA反馈。这种信息分享可提供正在开发的器械概况,特别是当未来6-12个月内有多个提交计划时,有助于FDA审查小组熟悉与目前可用器械有显著技术差异的新器械。虽然FDA工作人员可能会在信息交流会上提出一些需要澄清的问题,但他们通常只在会议期间听取信息,而不准备提供任何反馈。信息会议也可以用来记录FDA和提交者之间其他不属于Q-Submissions定义范围的互动。
E.其他Q-Submission类型 除了上述四种Q-Sub类型,Q-Sub计划还提供了一个机制来追踪FDA其他计划指南文件中描述的互动。目前,除上述Q-Sub类型外,Q-Submission计划中追踪的互动包括: PMA100天会议; FDA现代化法案下的协议和决定会议; 与突破性器械计划相关材料的提交; 与更安全技术计划(Safer Technologies Program, STeP)相关材料的提交; 附件分类请求。
F.Q-Submission计划其他使用情况 有一些互动不符合上述Q-Sub类型的定义,并且没有创建新的正式Q-Sub类型。当不存在新的Q-Sub类型用于追踪一个特定类型的互动时,FDA可使用信息会议Q-Sub类型作为追踪这些互动的工具。信息会议Q-Sub机制目前用于跟踪的互动类型的例子包括: 要求FDA对其他政府机构、非营利、贸易组织和专业协会的具体问题或交叉政策事项进行反馈。请注意,如果各组织在会议前自愿提交信息供FDA进行实质性审查,则提交的材料不是FDA与这些团体见面的必要条件,但FDA对接受这些材料持开放态度; 要求认可可公开访问的遗传变异数据库; 要求FDA对不属于Pre-Submission范围的临床研究设计要素进行反馈。这些请求可能包括关于NSR或IDE豁免研究的研究设计请求,其结果不打算支持未来的IDE或上市申请; FD&C Act 503(g)(2)节中定义的组合产品协议会议; 要求FDA反馈关于合规行为的问题。 总体而言,信息会议的目的是让提交者向 FDA 提供信息而不期望从 FDA 获得反馈。然而,当信息会议的 Q-Subs 被用来追踪,而正式的 Q-Sub 类型还没有被创建时,可以按照使用信息会议Q-Sub类型的计划规定提供反馈。
通过Q-Submission与FDA进行沟通是免费的,但不应因此将FDA当作咨询机构。医疗器械制造商应有技巧地提问,抓住核心,问题数量需符合要求,通过充分使用Q-Submission计划与FDA交流,提升器械注册成率。
参考资料:Requests for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program (fda.gov) 【供 稿】苏大检测医疗器械事业部法规中心
|




【本文地址】
今日新闻 |
推荐新闻 |