两分钟学会分析变异检测结果 – 云生信 |
您所在的位置:网站首页 › 测序结果文件的主要类型包括 › 两分钟学会分析变异检测结果 – 云生信 |
两分钟学会分析变异检测结果 – 云生信
|
上一次小果给大家详细地分享了变异检测的流程,这次为了让小伙伴们理解的更透彻,小果在这里为大家解读一下变异检测结果,废话不多说,我们直接开始吧: VCF文件FreeBayes生成的VCF(Variant Call Format)文件是一种常用的描述基因组变异信息的标准文件格式,用于描述SNP、INDEL和SV结果,这里小果以上次我们用FreeBayes处理得到的结果文件做如下说明:
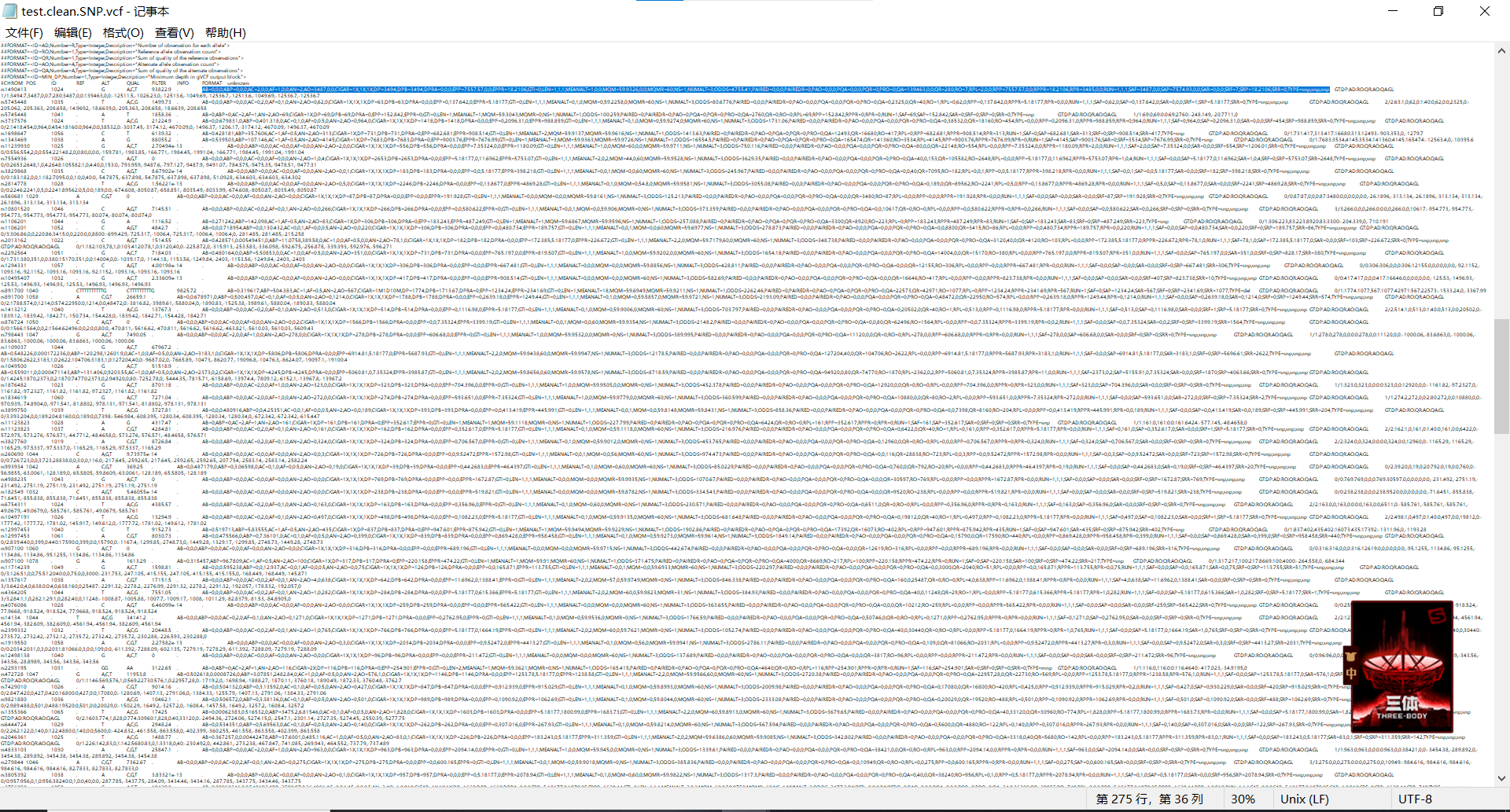
在VCF文件中,每一行表示一个位点的信息,列名和解释如下: CHROM:染色体名称,表示该位点所在的染色体。 POS:位点在染色体上的位置,以基因组坐标形式给出。 ID:位点的唯一标识符,可以是rs号(在dbSNP数据库中注册的SNP的标识符)或其他唯一ID。 REF:参考等位基因,表示在该位点上的参考序列。 ALT:备选等位基因,表示在该位点上可能存在的其他变异序列。 QUAL:质量得分,表示对该位点的置信度或可靠程度的估计。 FILTER:过滤状态,表示该位点是否通过特定的质量控制筛选。 INFO:附加信息,提供关于该位点的额外注释和相关信息,以键值对的形式呈现。 FORMAT:样本数据格式,指定在VCF文件的后续列中如何解释每个样本的基因型信息。 unknown:样本数据列,每个样本占据一列,表示对应位点的该样本的基因型和其他相关信息。小果提示:因为备忘录显示局限问题,INFO列内容太多,所以下图蓝色内容是INFO列的信息,显示大小放缩后的排列方式为本应的排列方式:
在VCF文件中,INFO列是用来注释每个变异记录的信息。其中,tag是INFO列中的一个字段,专门用来表示不同类型的突变。下面是不同标签的含义: SNP(Single Nucleotide Polymorphism):表示单核苷酸多态性,即在基因组中存在单个碱基发生替换的变异。 INS(Insertion):表示插入事件,即相对于参考基因组,在样本基因组中插入了一段新的碱基序列。 DEL(Deletion):表示缺失事件,即相对于参考基因组,样本基因组中删除了一段碱基序列。 MNP(Multiple Nucleotide Polymorphism):表示连续两个SNP位点的变异,例如,参考基因组中的AT被替换为样本基因组中的CG。 Complex(Composite Insertion and Substitution Events):表示复合插入和替换事件,即在样本基因组中同时发生了插入和替换的变异。根据上述描述,可以通过解析VCF文件并查找INFO列中特定标签的值来提取不同类型的突变记录。这样的分析有助于进一步研究和理解基因组变异在遗传学和疾病研究中的作用。 # 使用grep命令从freebayes.vcf文件中提取所有的SNP(单核苷酸多态性)记录,并将结果保存到新文件freebayes_snp.vcf中。 grep ‘TYPE=snp’ freebayes.vcf \> freebayes_snp.vcf INFO列还有两种信息比较重要,分别是: GT: 表示这个样本的基因型,对于一个二倍体生物,GT值表示的是这个样本在这个位点所携带的两个等位基因。0表示跟REF一样;1表示表示跟ALT一样;2表示第二个ALT。当只有一个ALT 等位基因的时候,0/0表示纯和且跟REF一致;0/1表示杂合,两个allele一个是ALT一个是REF;1/1表示纯和且都为ALT DP: 覆盖到这个位点的reads数量,相当于这个位点的深度(并不是所有的reads数量,而是达到一定质量值要求的reads数)。 限于篇幅,小果就不在这里对VCF文件中的INFO列的具体内容所代表的含义做具体解释啦,感兴趣的小伙伴们可以自行查阅哦~小果在这里给大家举例分享一下以下这列FORMAT列的信息:
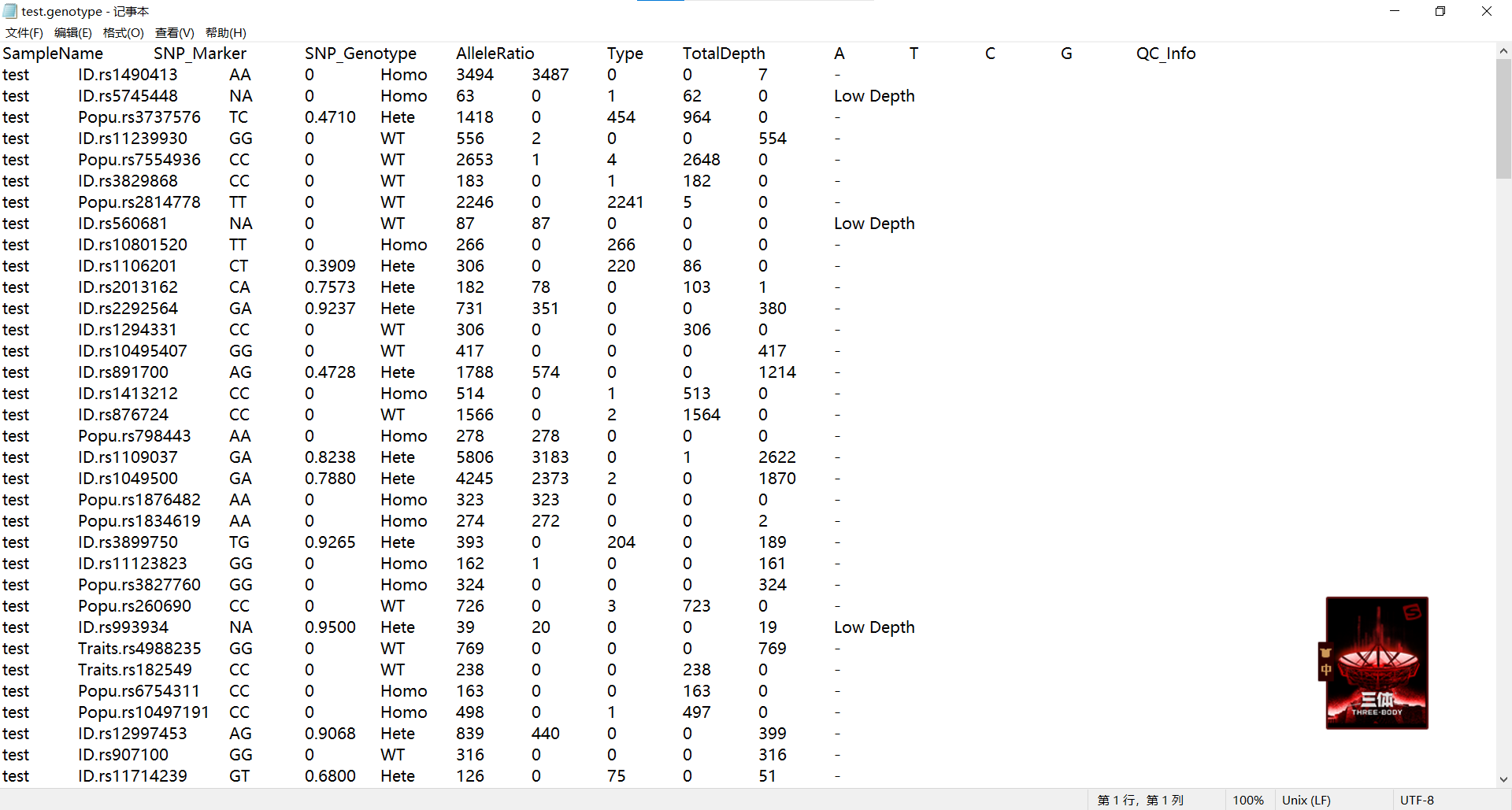
总结起来,VCF文件中的各列提供了关于每个位点的重要信息,包括位置、参考等位基因、备选等位基因、质量得分、过滤状态以及样本的基因型数据。通过解析这些列的内容,可以获取关于SNP和其他变异位点的详细信息。 genotype文件genotype文件是一种用于存储基因型数据的文件格式,这里小果以vcf2geno_free脚本处理所得的结果文件做介绍:
在genotype文件中,每一行表示一个样本在特定SNP位点的基因型信息,列名和解释如下: SampleName:样本名称或标识符,表示该行数据对应的样本。 SNP_Marker:SNP标记或位点编号,表示该行数据对应的SNP位点。 SNP_Genotype:基因型信息,表示该样本在该位点的基因型,通常由两个等位基因按照一定顺序组成。例如,AA表示纯合子(两个等位基因均为A),AB表示杂合子(一个等位基因为A,另一个为B)。 AlleleRatio:等位基因分离比例,表示等位基因A和B在该样本中的相对数量比例。 Type:基因型类型,表示该样本的基因型类型,如纯合子、杂合子或缺失。 TotalDepth:总深度,表示该位点在该样本中的测序深度,即用于该位点的测序读数总数。 A、T、C、G:各等位基因的深度,表示等位基因A、T、C和G在该样本中的测序深度。 QC_Info:质控信息,提供关于基因型质量控制的相关信息,可包括SNP位点的质量得分、过滤状态等。综合起来,genotype文件中的各列提供了每个样本在特定SNP位点上的基因型信息和相关测序统计数据。这些信息可以用于研究和分析不同样本之间的遗传变异、基因型频率以及其他与等位基因相关的特征。
以第一行的样本为例,样本名称为test,SNP标记为ID.rs1490413,基因型为AA,等位基因比例为0,基因型类型为纯合型(Homo),总深度为3494,A等位基因深度为3487,T等位基因深度为0,C等位基因深度为0,G等位基因深度为0,质控信息为7,基因质量值为-。 怎么样,是不是非常详尽,小伙伴们都收获匪浅呢?今天的分享就到这里啦,如果小伙伴们平时在生信分析的操作过程中遇到困难,欢迎大家使用小果开发的生信工具平台http://www.biocloudservice.com/home.html哦。 今天的分享就到这里啦,下一次小果会给大家带来详尽的变异检测原理,想要提升自己、成长为生信大神的小伙伴们一定不要错过哦,那么我们下次再见啦,拜拜~ |




【本文地址】
今日新闻 |
推荐新闻 |