提取 VCF 文件中的基因型信息 |
您所在的位置:网站首页 › 如何提取基因组一个基因序列 › 提取 VCF 文件中的基因型信息 |
提取 VCF 文件中的基因型信息
|
VCF 是一种常用的生物信息学文件格式,用于存储基因组中的遗传变异数据,特别是单核苷酸多态性(SNP)和小插入/缺失(Indel)等变异。 VCF 文件通常由文本格式组成,可以用文本编辑器进行查看和编辑。每个 VCF 文件通常包含多行记录,每一行表示一个位点上的变异。这些记录包含了位点的染色体位置、参考基因型、变异的基因型信息以及相关的质量控制指标等。
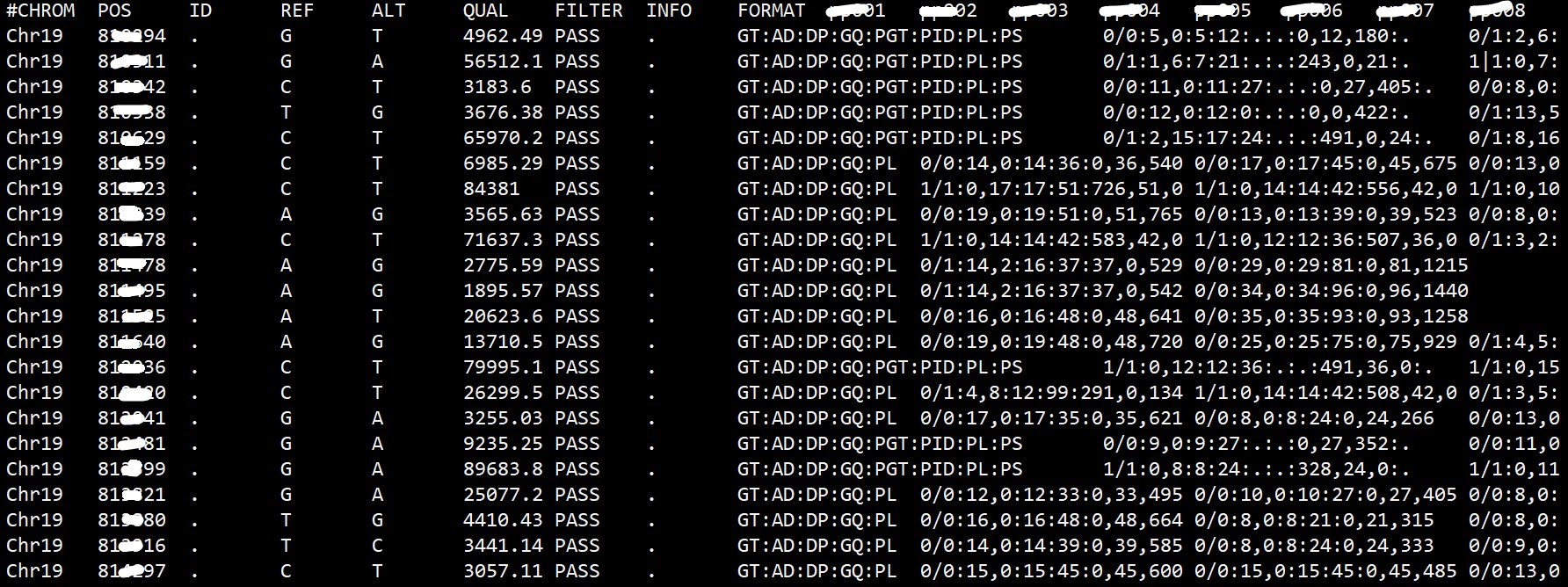
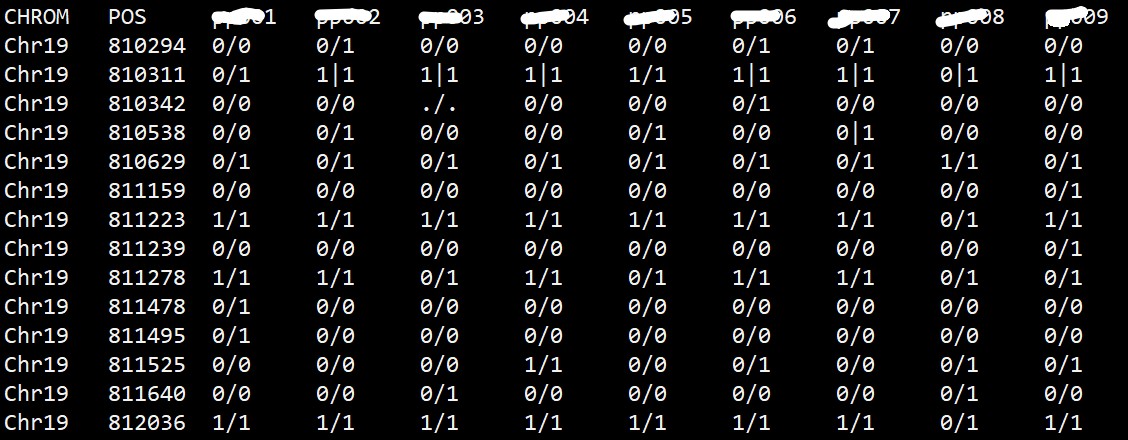
VCF 文件中的基因型信息可以用不同的方式表示,常见的有基于基因型(genotype)的表示和基于等位基因(allele)的表示。基于基因型的表示方式使用字母和数字的组合表示不同的基因型,如AA、AT、CC、0/1、1/2 等。基于等位基因的表示方式使用字母表示等位基因,如A、T、C、G 等。 目的:想提取vcf文件的基因型信息,生成下图的格式
有许多软件可以用来提取 VCF 文件中的基因型信息。以下是一些常用的软件工具: 1.bcftoolsbcftools 是一组用于操作 VCF 文件的命令行工具,它是 samtools 软件包的一部分。你可以使用 bcftools 的 query 命令来提取 VCF 文件中的基因型信息。例如,使用以下命令可以提取所有样本的基因型: bcftools query -f '%GT\n' your_file.vcf还可以根据需要自定义输出格式。 2.VCFtoolsVCFtools 是一个流行的 VCF 文件处理工具集,它提供了许多功能,包括提取基因型信息。你可以使用 --extract-FORMAT-info 选项来提取基因型信息。例如,使用以下命令可以提取所有样本的基因型: vcftools --vcf your_file.vcf --extract-FORMAT-info GT --out output_file 这将生成一个包含基因型信息的新文件。3.GATK (Genome Analysis Toolkit)GATK 提供了丰富的工具和库用于基因组数据分析。它包含了用于处理 VCF 文件的工具,可以提取基因型信息。你可以使用 GATK 的 VariantsToTable 工具来提取基因型。例如,使用以下命令可以提取所有样本的基因型:gatk VariantsToTable -V your_file.vcf -F GT -O output_table.txt这将生成一个包含基因型信息的表格文件。 这只是一小部分可用的工具,还有其他许多软件和编程库可用于提取 VCF 文件中的基因型信息。你可以根据自己的需求选择适合的工具,并根据具体的软件文档了解更多使用方法和选项。 |
【本文地址】