向酿酒酵母中导入线性质粒的方法及制备的酵母菌与流程 |
您所在的位置:网站首页 › 如何把质粒导入酵母菌中 › 向酿酒酵母中导入线性质粒的方法及制备的酵母菌与流程 |
向酿酒酵母中导入线性质粒的方法及制备的酵母菌与流程
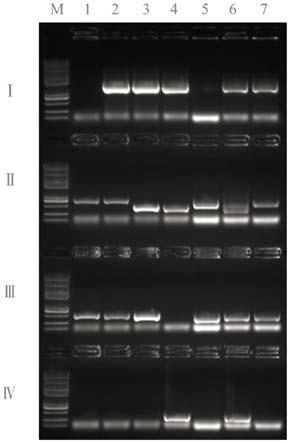 [0001]本申请涉及分子生物学技术领域,具体而言,涉及一种向酿酒酵母中导入线性质粒的方法及制备的酵母菌。 背景技术: [0002]在定向进化过程中,对生物分子进行重新改造以寻找发挥着有益新功能的新型生物分子。这一技术正在彻底改变药物开发、化学工程和其他应用。近年开发的两种定向进化方法page(esvelt,kevin,m,et al.a system for the continuous directed evolution of biomolecules.[j].nature,2011)和orthrep(ravikumar a,arrieta a,liu c c.an orthogonal dna replication system in yeast.[j].nature chemical biology,2014,10(3):175-7)简化了定向进化流程,减少了实验室工作,提高了定向进化效率。[0003]orthrep定向进化的原理是乳酸克鲁维酵母(kluyveromyceslactis)细胞质中存在两个线性质粒pgkl1(p1)和pgkl2(p2),p1和p2的复制用的是各自线性质粒上编码的一个独立的dna聚合酶,这两个dna聚合酶不能复制核基因组dna和其他质粒dna,反之,复制核基因组dna和其他质粒dna的dna聚合酶也不能复制p1和p2。这是两套完全不相关的dna复制系统。将负责复制p1的dna聚合酶(tp-dnap1)突变,提高复制p1时错误碱基的掺入率,同时根据酵母核基因密码子的偏好性,将编码tp-dnap1突变体的基因密码子优化后,在酵母细胞核中进行表达,进而产生突变的tp-dnap1。该突变的tp-dnap1在复制p1质粒时,可高频率的掺入错误碱基,从而使p1质粒上的基因高频率的突变。[0004]应用orthrep定向进化系统已成功进化出抗药性疟疾二氢叶酸还原酶(arjunravikumar,et al.scalable,continuous evolution of genesat mutation rates above genomic errorthresholds.[j].cell,2018)。目前,还未见应用orthrep定向进化系统进化互作蛋白对的报道。[0005]用于研究互作蛋白对最多的酵母菌株为商业化菌株酿酒酵母ah109(mata,trp1-901,leu2-3,112,ura3-52,his3-200,gal4δ,gal80δ,lys2::gal1uas-gal1tata-his3,mel1 gal2uas-gal2tataade2,ura3::mel1uas-mel1tata-lacz)。该菌株遗传背景清楚,已多年用于研究互作蛋白对,积累了丰富的经验,且与该菌株配套的试剂较容易购买。为了建立进化蛋白对的orthrep定向进化系统,需要向商业化菌株酿酒酵母ah109等目标酿酒酵母中导入p1和p2质粒。向酵母导入p1和p2的方法已有报道,如原生质体融合法(gunge n,sakaguchi k.intergeneric transfer of deoxyribonucleic acid killer plasmids,pgkl1 and pgkl2,from kluyveromyceslactis into saccharomyces cerevisiae by cell fusion.[j].journal of bacteriology,1981,147(1):155)、纯化的p1和p2线性质粒直接导入原生质体法(gunge n,murata k,sakaguchi k.transformation of saccharomyces cerevisiae with linear dna killer plasmids from kluyveromyceslactis.[j].journal of bacteriology,1982,151(1):462-4)。这两种方法均用到原生质体,操作繁琐,且效率低下。目前还没有一种简单、高效、高质量的向酿酒酵母导入p1和p2的方法。 技术实现要素: [0006]本发明提供了一种向酿酒酵母中导入线性质粒的方法,该方法包括向源酿酒酵母的线性质粒p1中整合筛选标记基因,并使酵母交配时负责核融合的kar1基因无法行使核融合功能,由此得到改造后的源酿酒酵母;经改造后的源酿酒酵母与目标酿酒酵母交配,使线性质粒p1和p2导入目标酿酒酵母菌株中。当源酿酒酵母与目标酿酒酵母的交配型相同时,上述方法还包括交配前,先转换改造后的源酿酒酵母的交配型。[0007]具体的,该方法包括下述(1)-(5)中的一个或多个:[0008](1)所述源酿酒酵母中存在p1和p2两个线性质粒;[0009](2)所述筛选标记基因为目标酵母菌株的缺陷基因;[0010](3)所述目标酿酒酵母中不存在线性质粒p1或p2;[0011](4)所述使酵母交配时负责核融合的kar1基因无法行使核融合功能是指删除kar1基因的部分序列,具体的是指敲掉kar1基因编码区289-573间共285个碱基;[0012](5)所述转换改造后的源酿酒酵母的交配型是根据实际需要,通过同源重组的方式将matα或mata基因序列导入改造后的源酿酒酵母菌株中。[0013]具体的,所述源酿酒酵母可以为ga-y233菌株,该菌株由酿酒酵母f102-2菌株经ura3基因和trp1基因失活处理而得,所述酿酒酵母f102-2菌株来源于全球生物资源中心(american type culturecollection,po box 1549,manassas,va 20108usa),保存目录号为200585;所述目标酿酒酵母为ah109菌株;所述方法具体包括:[0014](1)在源酿酒酵母ga-y233菌株的p1质粒上整合trp1筛选标记基因,便于交配后含有p1和p2质粒的ah109菌株的筛选;[0015](2)鉴于kar1基因突变的酵母菌株不能进行细胞核融合,用crispr-cas9介导同源重组的方法敲掉kar1基因编码区289-573间共285个碱基,便于两酵母菌株交配后细胞质融合而细胞核不融合;[0016](3)鉴于酿酒酵母ga-y233和酿酒酵母ah109交配型均为mata,两菌株不能进行交配,克隆matα基因序列,并用crispr-cas9介导同源重组的方法将步骤(2)获得的改造后的源酿酒酵母ga-y233的交配型修改为matα,便于酿酒酵母ga-y233和酿酒酵母ah109的交配;[0017](4)将步骤(3)获得的改造后的酿酒酵母ga-y233菌株和目标酿酒酵母ah109交配后,在不含尿嘧啶和色氨酸的缺陷型培养基上培养,pcr筛选含有p1和p2质粒的ah109菌株。[0018]进一步的,所述方法包括下述(a)-(g)中的一个或多个:[0019](a)所述步骤(1)中整合的trp1筛选标记基因编码序列如seq id no.2所示;[0020](b)所述步骤(1)中检测所用引物对为f-p1-trp和r-p1-trp,其序列分别如seq id no.4和5所示;[0021](c)所述步骤(2)中基因重组采用的靶位点为kar1基因的编辑载体pcas-kar1的序列如seq id no.13所示;[0022](d)所述步骤(2)中检测所用引物对为f-kar1-jc和r-kar1-jc,其序列分别如seq id no.14和15所示;[0023](e)所述步骤(3)中基因重组采用的靶位点为mata基因的编辑载体pcas-mata的序列如seq id no.22所示;[0024](f)所述步骤(3)中检测所用引物为f-α-jc、f-a-jc和r-α/a-jc,其序列分别如seq id no.23-25所示;[0025](g)所述步骤(4)中检测所用引物对为f-p1-trp和r-p1-trp、f-kar1-jc和r-kar1-jc、f-α-jc和r-α/a-jc、f-a-jc和r-α/a-jc。[0026]本发明还提供一种酿酒酵母重组工程菌,该酿酒酵母重组工程菌采用前述的方法制备而成。[0027]本发明还提供一种酿酒酵母重组工程菌,该酿酒酵母重组工程菌以ga-y233菌株为出发菌株,在ga-y233菌株的p1质粒上整合有trp1筛选标记基因;且该酿酒酵母重组工程菌的kar1基因不能行使核融合功能,具体的通过敲掉kar1基因编码区289-573间共285个碱基使其不能行使核融合功能;其中,所述ga-y233菌株由酿酒酵母f102-2菌株经ura3基因和trp1基因失活处理而得,所述酿酒酵母f102-2菌株来源于全球生物资源中心(american type culturecollection,po box 1549,manassas,va20108usa),保存目录号为200585。[0028]进一步的所述酿酒酵母重组工程菌的交配型为matα型。具体的,通过克隆matα基因序列,并用crispr-cas9介导同源重组的方法将所述酿酒酵母重组工程菌的交配型修改为matα。[0029]本发明还提供一种前述的方法和采用前述方法制备的酿酒酵母重组工程菌在定向进化领域的应用。具体的,该应用为互作蛋白对的定向进化领域。[0030]本发明的有益效果包括:本发明提供的向酿酒酵母中导入线性质粒的方法,其操作简单,且能够高效、高质量的向酿酒酵母中导入线性质粒,经pcr检测,导入成功率可达95%以上。附图说明[0031]图1为酿酒酵母菌株pcr鉴定电泳图,其中第i行检测所用引物为f-p1-trp和r-p1-trp;第ⅱ行检测所用引物为f-kar1-jc和r-kar1-jc;第ⅲ行检测所用引物为f-a-jc和r-α/a-jc;第ⅳ行检测所用引物为f-α-jc和r-α/a-jc;样品m为trans2k plusⅱdna marker;样品1为ga-y233;样品2为ga-y233-trp;样品3为ga-y233-trp-kar1;样品4为ga-y233-trp-kar1-matα;样品5为ah109;样品6为ah109×ga-y233;样品7为ah109-p1-trp。具体实施方式[0032]下面结合实施例对本发明进行进一步说明和描述,但所描述的实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明和实施例中,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他发明和实施例,都属于本发明保护的范围。[0033]若未特别指明,实施例均按照常规实验条件,如sambrook等编写的分子克隆实验手册(new york:gold spring harbor laboratory press,1989)、clontech编写的酵母操作指南(pt3024-1,2009)、或按照制造厂商说明书建议的条件。[0034]下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。[0035]实施例1、向酿酒酵母ah109中导入线性质粒的方法[0036]包括如下步骤:kar1-jc:ggatctgattcagcacattg(seq id no.14)和r-kar1-jc:gaagtctatgccatttagac(seq id no.15)。若kar1基因未发生重组,pcr产物为817bp,具体序列见seq id no.16;若kar1基因发生了重组,pcr产物为532bp,具体序列见seq id no.17。[0048]将kar1基因发生了重组的ga-y233-trp酵母菌株命名为ga-y233-trp-kar1。该菌株可与交配型为matα的酵母菌株交配,向后者导入线性质粒p1-trp和p2,交配后细胞质融合而细胞核不融合。[0049](3)酿酒酵母ga-y233-trp-kar1菌株的交配型转换为matα[0050]在sgd数据库(https://www.yeastgenome.org/)查询matα基因序列。查到s288c菌株的matα基因的具体序列见seq id no.18。根据该序列设计引物f-α:gaattcttgtattagacgagggacggagtg(seq id no.26)和r-α:[0051]gaattcttgattgtttgcttgagtctgag(seq id no.27)。用上述引物对以酿酒酵母ga-y233基因组dna为模板进行pcr扩增,获得2519bp片段。该片段两端为ecorⅰ酶切位点,与seq id no.18比对发现有7个碱基的差异,具体序列见seq id no.19。[0052]根据sgd数据库中查到的s288c菌株mata基因的具体序列(seq id no.20)和该基因的具体标注信息,设计crispr-cas9的靶位点序列为acaaaaatatttctaacaat(seq id no.21),将该序列插入酵母基因组编辑载体pcas,获得靶位点为mata基因的编辑载体pcas-mata,具体序列见seq id no.22。[0053]将ecorⅰ酶切回收的2519bp片段matα基因片段和pcas-mata质粒混合,共转化酿酒酵母ga-y233-trp-kar1感受态细胞。转化细胞涂布在含有100mg/l g418的ypda培养基上培养。72h后将长出的克隆在不含g418的ypda培养基上划线,继续生长36h。将划线的克隆做pcr检测,所用引物为f-α-jc:gcacggaatatgggactacttcg(seq id no.23)、f-a-jc:actccacttcaagtaagagtttg(seq id no.24)和r-α/a-jc:agtcacatcaagatcgtttatgg(seq id no.25)。f-α-jc和r-α/a-jc扩增片为404bp,f-a-jc和r-α/a-jc扩增片段为544bp。若扩增出404bp片段,表明该菌株的交配型为matα;若扩增出544bp片段,表明该菌株的交配型为mata。若同时扩增出404bp片段和544bp片段,表明该菌株为二倍体。[0054]将交配型转换为matα的ga-y233-trp-kar1酵母菌株命名为ga-y233-trp-kar1-matα。该菌株可与商业化酵母菌株ah109交配,向后者导入线性质粒p1-trp和p2。[0055](4)用ga-y233-trp-kar1-matα酵母菌株向ah109酵母菌株导入p1-trp和p2[0056]挑取ypda平板上生长的ga-y233-trp-kar1-matα酵母菌株克隆和ah109酵母菌株克隆放入盛有5ml ypda液体培养基的50ml试管,摇菌至吸光值od600到0.5-0.6。将两种酵母菌液1:1混合,将混合菌液用移液枪一滴滴的滴在ypda平板上。30℃培养5h,用缺少色氨酸和尿嘧啶的缺陷型液体培养(sc-trp-ura)洗脱,将洗脱液涂布在sc-trp-ura平板上,30℃培养96h后平板上出现单克隆。[0057]挑取sc-trp-ura平板上长出的酵母单克隆,在sc-trp-ura平板上划线,生长36h后,pcr检测克隆的基因型。检测的酵母克隆分为两类:一是含有p1-trp和p2质粒的ah109菌株,命名为ah109-p1-trp;另一类为ah109菌株和ga-y233-trp-kar1-matα菌株杂交产生的二倍体,命名为ah109×ga-y233。检测所用的引物对为:f-p1-trp和r-p1-trp、f-kar1-jc和r-kar1-jc、f-α-jc和r-α/a-jc、f-a-jc和r-α/a-jc。如附图1所示,ah109-p1-trp菌株的扩增基因型为:f-p1-trp和r-p1-trp pcr扩增产生1554bp片段;f-kar1-jc和r-kar1-jc pcr扩增产生817bp片段;f-α-jc和r-α/a-jc pcr扩增不出片段,f-a-jc和r-α/a-jc扩增产生544bp片段。ah109×ga-y233二倍体菌株的扩增基因型为:f-p1-trp和r-p1-trp pcr扩增产生1554bp片段;f-kar1-jc和r-kar1-jc pcr扩增产生817bp片段和532bp片段;f-α-jc和r-α/a-jc pcr扩增产生404bp片段,f-a-jc和r-α/a-jc扩增产生544bp片段。[0058]经上述四对引物pcr检测,生长的酵母克隆多为ah109-p1-trp菌株,占比95%以上。 |
【本文地址】