细胞和基因治疗类产品开发要点(CMC篇) |
您所在的位置:网站首页 › 化学cmc › 细胞和基因治疗类产品开发要点(CMC篇) |
细胞和基因治疗类产品开发要点(CMC篇)
|
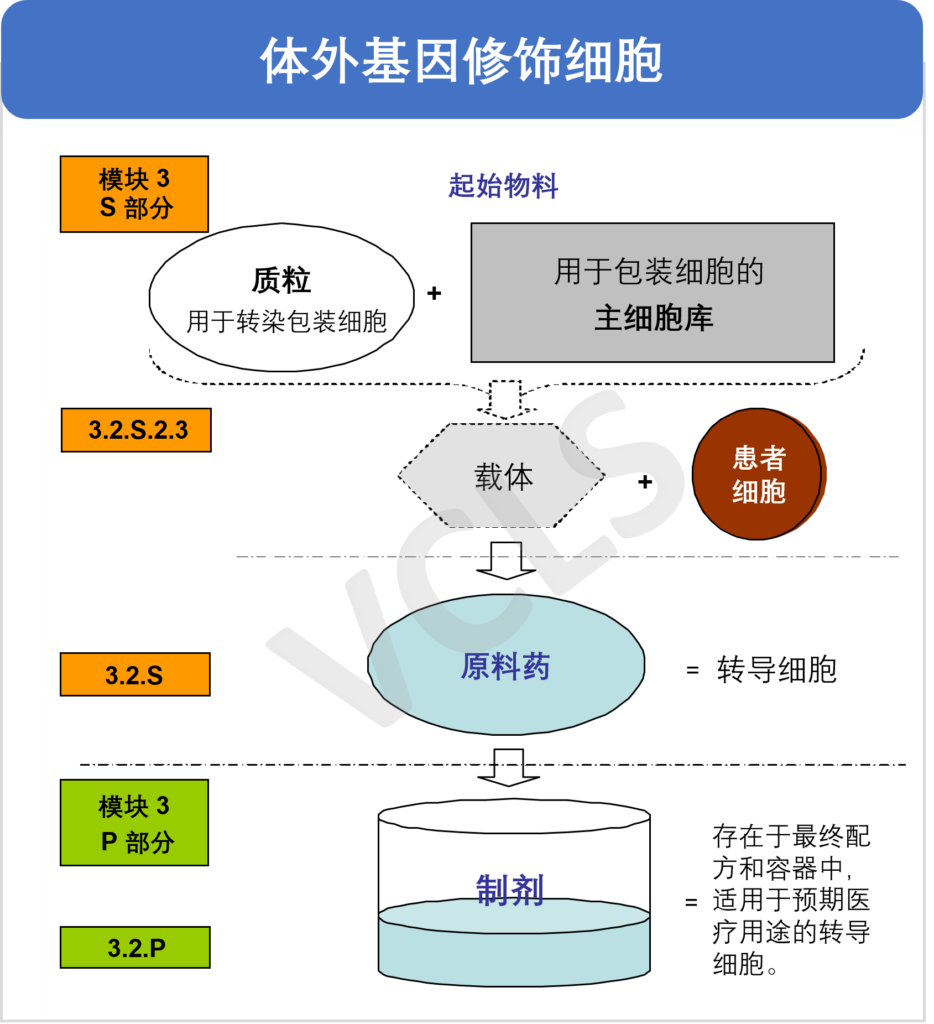
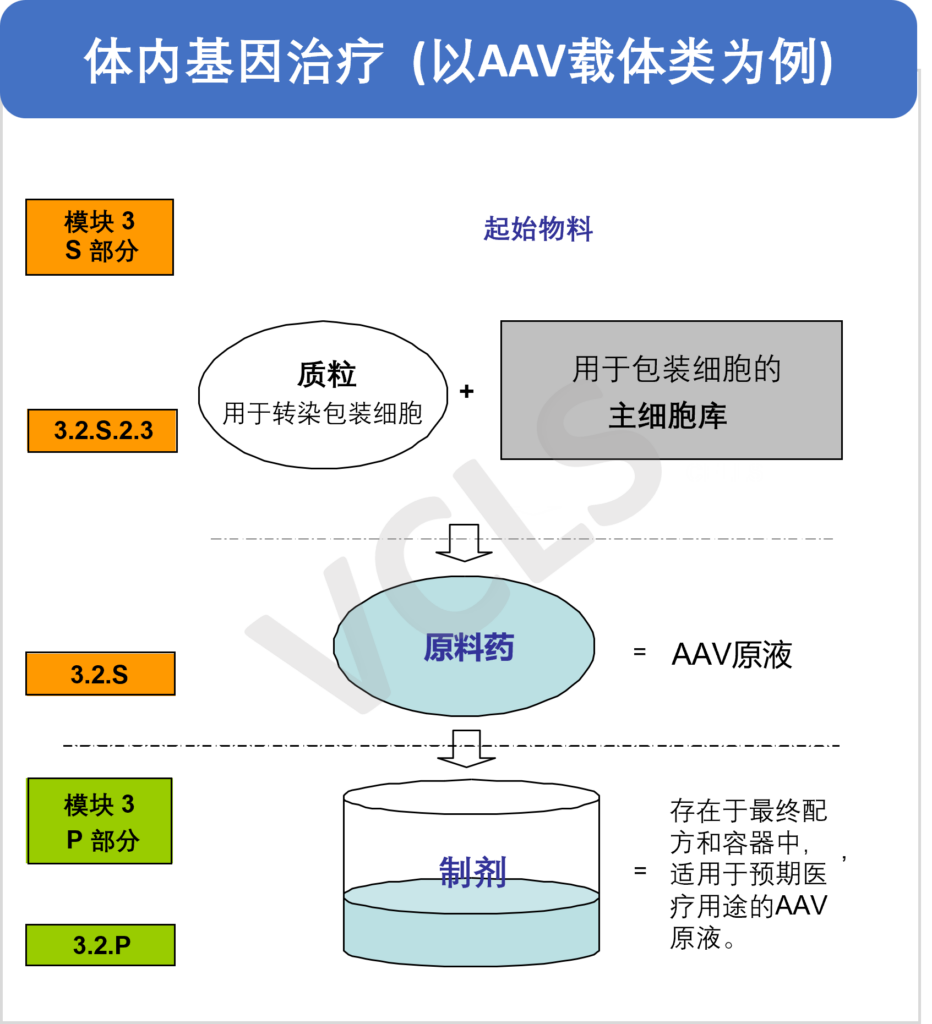
细胞和基因治疗产品都面临着特定的CMC(化学、生产和控制)的挑战,而这些挑战对于此类产品是否能够最终获批上市起到了决定性作用。在不久前刚刚结束的线上研讨会中, VCLS的药政专家Jennifer Woods博士针对此类产品的CMC开发要点进行了分享。本篇文章将对该部分内容进行回顾与总结。 原材料、起始物料、原料药及制剂的界定相比小分子药物和传统的生物制品,应用于细胞和基因治疗产品中物料的术语往往并不明确而且相当复杂。不同地区的监管部门对其定义也尚未统一。关于该部分,您也可以参考我们的“细胞和基因治疗制品中物料的界定和考量”系列推文,获取更多相关信息。 无论是原材料,辅助物料还是起始物料,都必须保证适当的质量,因为材料中存在的任何杂质最终都可能留存于最终产品中。所有物料都必须考虑到他们是否有将病毒污染物引入工艺流程的风险。生产者应尽可能避免使用动物源性材料,并使用在符合GMP要求的材料,以确保质量和有效的质控。 然而对于细胞和基因疗法产品,很多时候仅有研究级原材料和起始物料可用。在此类情况下,我们建议开发者根据GMP的相关原则采用基于风险的方法来组织生产,并充分评估使用该研究级材料的风险。这些风险不仅包括物料中潜在杂质的风险,还包括在大量生产时能否持续进行供应的风险。此外,应确保这些原材料针对预期用途的适用性,包括是否适用于测试(例如生物学性能和安全性测试)。 对于开发者而言,确保所使用的所有材料都具有合规的质量是重中之重。 起始物料的差异性起始物料的差异性性对所有细胞和基因治疗产品而言都是个问题,但是基于细胞的治疗产品,尤其是自体性产品,在细胞起始物料上有显著的差异。自体性产品是指那些从患者身上收集细胞作为起始材料的治疗方法。由于患者的健康状况、年龄和疾病状况各不相同,其身上的细胞产量和健康状况也会有很大的差异。即使是异体细胞疗法,也会依赖于捐献者社群。他们容易受到捐助的不一致性和捐助者多样性不足等问题的影响。因此,了解COA之外的原料和起始物料的质量属性是很重要的。产品的生产过程和产品(或产品的一个组分)可能会以一种在进行COA分析时没有考虑到的方式影响您的起始物料。 此外,随着生产规模的扩大,物料的性质通常会随之产生变化,在工艺放大时降低这方面的风险十分重要。我们也建议对原材料按其关键程度进行分类,其中血清、培养基、生长因子和细胞因子是最关键的。如果条件允许,生产者应该在产品开发早期就进行稳健性研究,使用不同批次的起始物料和原材料,以帮助识别不能从COA中发现但影响产品质量的物料属性。 组分界定对CTD模块3的影响对于细胞和基因疗法来说,原料和起始物料、原料药和制剂之间的划分并不是直截了当的。其定位的选择,不仅会影响CMC部分内容材料的撰写,还会影响所选择的控制策略。只有在正确定义这些组分的前提下,原料和起始物料、原料药和制剂的控制策略才会更容易制定和证明恰当,包括放行检测、起始物料检验、原料检验、过程控制和病毒安全性风险评估。 以下为模块3中针对不同CGT产品的示例。图1是一个体外基因修饰的细胞产品。起始物料包括质粒(用于转染包装细胞)以及主细胞库(包装细胞)。这两部分能够生产出用于转导患者细胞的载体。所有这些组份都需要在模块3的S.2.3材料控制部分中定义,包括在该过程中使用的任何原材料。原料药则是被转导的患者的细胞。在细胞类产品中,原料药通常被立即处理为最终的制剂产品,即存在于最终配方和包装容器中。 图2是体内基因治疗的产品,以腺相关病毒(AAV)制剂为例。其起始物料包括用于生产AAV的质粒和主细胞库。原料药为纯化AAV原液。在AAV类产品,原料药通常会在加工为制剂前储存一段时间,可能在不同的设施中进行储存。制剂是在最终产品容器中经过无菌过滤、配制的AAV。
对于细胞和基因疗法相关产品,其最初的开发通常是在学术实验室环境下进行的,通常会在实验室规模下进行开发并使用研究级的材料。通过这个过程获得的产品通常仅能用于概念验证研究和分析方法的开发,也可用于人体首次试验所支持的非临床研究。为了符合开展人体首次试验的要求,产品必须严格按照cGMP中的要求进行生产。很多生物技术企业往往选择和CDMO合作,包括后期生产过程和分析方法的转移。然后随着工艺的转移,开发者往往需要进行进一步的CMC开发,包括适应相应的厂房和设施,物料变更,工艺变更以减少杂质或增大产量/规模,或开发新的分析方法等部分。 产品初步开发方案(移交CDMO前)在向CDMO进行技术转移之前,开发者应收集所有可用的标准操作规程、出版物、概念验证研究和产品表征数据,并准备“知识转移包”,定义生产工艺、批量、分析材料包、物料清单和生产设备。开发者需要从此处就开始与CDMO合作,根据其生产设施和能力进行相应调整。此外,开发者还应列出包括关键原料在内的制造工艺之间的所有差异,以便于未来讨论不同工艺的产品的可比性。 随着开发的进行,可比性研究会变得越来越昂贵和复杂。因此,提前预测后期需求,甚至提前进行产业化无疑是具有相当大的好处的,其能够使得开发者最大限度地减少关键非临床研究或早期临床阶段产品与已上市产品之间的潜在差异。例如,随着临床阶段的进展,开发者可能需要减少杂质,例如工艺残留,不需要的细胞类型或空的AAV衣壳。凡是需要进行变更,都必须进行相应的风险评估,以及可能需要的可比性研究。 可比性研究有时候尽管对开发末期和商业化的需求进行了预期,但在开发过程中仍然不可避免地需要进行一些变更。可比性研究的步骤包含:定义产品的关键质量属性,确定相关的分析方法,通过适当的研究评价产品相对于预期变化的稳健性,最后,实施变更并评估对产品的影响。可比性研究的结果是非常重要的,如果没有从质量角度证明可比性,一些非临床甚至临床研究可能需要进行桥接。 监管机构期望开发者能够按照具有预先确定标准的方案进行可比性研究。开发者应向其证明其应用于评估变更前和变更后产品情况的检测方法是合适的。对于重大变更,通常需要进行加速稳定性或强制性降解研究作为可比性的一部分,以确保该变更不会影响产品的稳定性或随着时间的推移可能形成的降解物。我们建议开发者与监管机构针对拟议的可比性研究策略进行讨论,特别是在产品开发的晚期阶段,以获得其对研究所需批次数量以及可接受标准的指导意见。 早期产品表征与初代质量标准制定细胞和基因疗法的表征和检测是十分复杂的。早期开发批次的样品数量通常相当有限,有些产品不能冷冻保存需要快速进行检测,开发过程中产品往往缺乏现成的可用标准,对其产品作用机制的理解也可能是并不完整。即使了解其完整作用机制也常常不能通过合适的分析检测重现或者代表。此外,由于许多细胞和基因治疗项目的开发时间线非常紧张,导致产品的分析开发时间往往是捉襟见肘的。同时分析方法还高度依赖于产品本身。因此在缺乏用于此类产品的通用方法的情况下,开发者应直接基于产品的表征研究仔细考量,并且应尽最大可能基于产品知识制定适用的检测策略,并整合可用的非临床甚至临床研究的数据。基于过往的经验,我们发现这种集合CMC/非临床/临床综合考量的方式(integrated approach)对细胞和基因治疗产品分析开发非常有效,这其中又以作用机制以及效力测定作为核心关注点。 效力测定方法效力是产品表征的关键属性,由于产品的复杂性,大多数产品需要采用矩阵方法进行效力测定,包括直接测定和间接测定。MoA中的每一个要素都可以单独用于开发一系列效力测定方案,这些检测可以在整个临床开发过程中进行逐步实施。 生物学活性的直接测定是指基于产品特定属性的体外或体内生物学方法。生物学活性的间接测定是一种替代性测定方法。它是一种基于与生物学活性相关的产品属性(如免疫化学、分子、物理或生化特性)的非生物测定分析方法。 在为体内基因疗法开发效力测定方法时,我们推荐分两大步骤。首先,载体必须将遗传物质引入细胞;其次,引入的物质必须在转导细胞中产生预期的生物效应。 感染滴度、载体基因组滴度和衣壳滴度常被作为效力测定矩阵的一部分,但它们不能证明产品的全部功能。靶细胞(或代表性细胞)的转染效率是MOA的第一步。通常在I期研究中,转染效率测定结合滴度测定就足以确定产品的效力基质。 随着产品开发进程的深入,开发者必须展示更多的MOA。对其而言,下一步是研究靶细胞中的基因表达和功能。这部分可能难以建模,但至少需要明确: 替代指标与体内生物学功能相关,并提供了科学依据 选择用于效力测定的细胞类型能够代表体内靶细胞 实验条件能够代表体内转导过程值得关注的是,欧盟和美国对早期阶段对效力测定的要求各不相同,FDA要求在Phase3临床试验前完成效力测定的开发并在3期完成验证,在Phase1阶段甚至可以进行非定量检测,EMA则要求相对较高,希望在早期阶段看到一个较为完整的产品特性分析框架,其中包括定量效力测定。 我们建议产品开发者在开发过程中与监管部门密切合作,将CMC、非临床和临床开发协同进行,时刻注意并参考最新的监管指导文件、技术以及方法,并采用基于风险的方法来确定哪些研究/测试是必要的,以确保项目在各个方面的连续性和合规性。
发布于: 2023年8月17日
|
【本文地址】
今日新闻 |
推荐新闻 |