samtools 统计重测序数据深度 depth、depth |
您所在的位置:网站首页 › samtools参数 › samtools 统计重测序数据深度 depth、depth |
samtools 统计重测序数据深度 depth、depth
|
测试数据链接: 链接:https://pan.baidu.com/s/18_R3W1K7DYdUA9DxGZ0jxw 提取码:nklh --来自百度网盘超级会员V6的分享
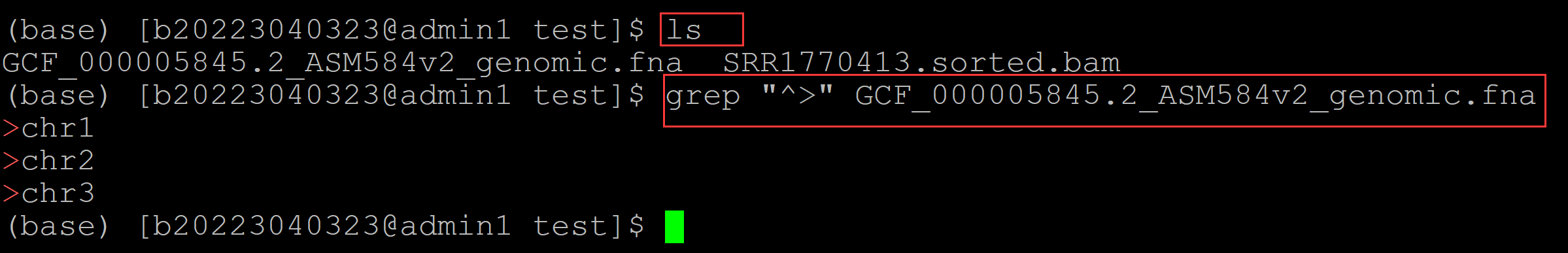
背景知识: 将质控后的fastq文件比对到参考基因组之后,有可能会有部分染色体根本比对不上。 比如参考基因组中一共有10条染色体,结果fastq的reads仅比对到8条染色体,那么就是说有两条染色体压根就没有比对上。 用一个实际的测试数据来看一下: 这里准备了两个文件:一个参考基因组fasta文件,和一个按照一般流程将fastq的reads数据比对到参考基因组后生成的排序后的bam文件,利用这两个文件可以查看参考基因组一共有多少条染色体, fastq数据一共比对上多少条染色体。 统计参考基因组的染色体数目: (base) [b20223040323@admin1 test]$ ls GCF_000005845.2_ASM584v2_genomic.fna SRR1770413.sorted.bam (base) [b20223040323@admin1 test]$ grep "^>" GCF_000005845.2_ASM584v2_genomic.fna ## 统计参考基因组的染色体数目 >chr1 >chr2 >chr3
可见该参考基因组一共有三条染色体。
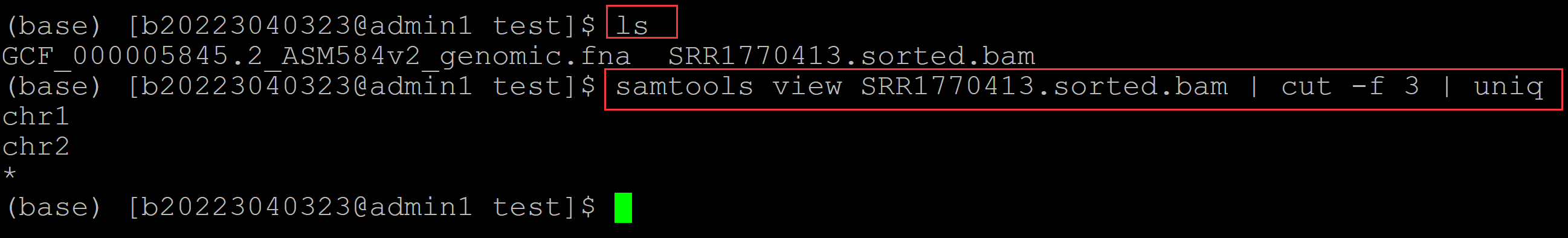
利用bam文件统计一共比对上多少条染色体: (base) [b20223040323@admin1 test]$ ls GCF_000005845.2_ASM584v2_genomic.fna SRR1770413.sorted.bam (base) [b20223040323@admin1 test]$ samtools view SRR1770413.sorted.bam | cut -f 3 | uniq ## 利用bam文件统计一共比对上多少条染色体 chr1 chr2 *
可见一共比对上的染色体数有两条,也就是说chr3并没有比对上。
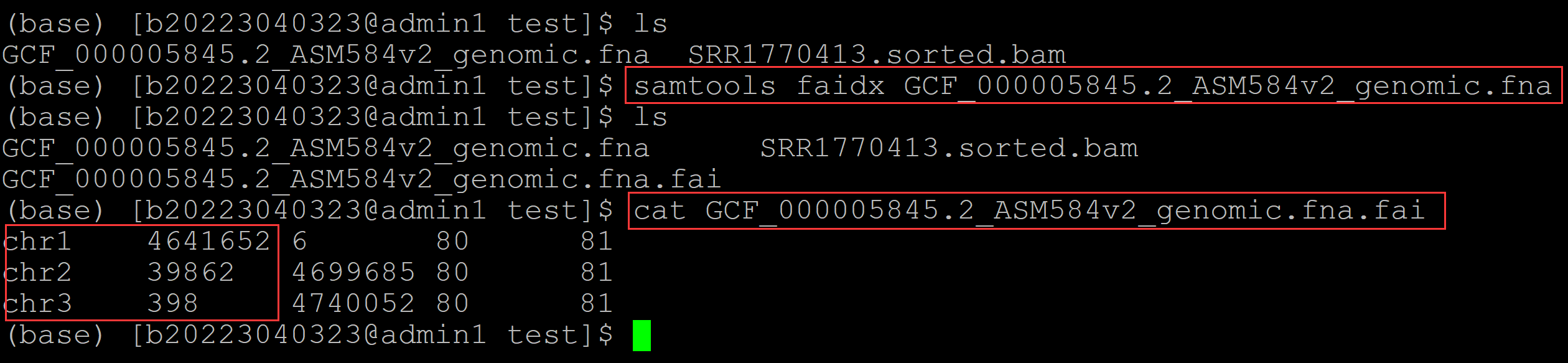
比较samtools计算测序深度时:depth、depth -a、depth -aa的区别。 001、首先先统计一下参考基因组中每一条染色体的长度,可以使用samtools软件实现: (base) [b20223040323@admin1 test]$ ls GCF_000005845.2_ASM584v2_genomic.fna SRR1770413.sorted.bam (base) [b20223040323@admin1 test]$ samtools faidx GCF_000005845.2_ASM584v2_genomic.fna ### 统计每一条染色体的长度 (base) [b20223040323@admin1 test]$ ls GCF_000005845.2_ASM584v2_genomic.fna SRR1770413.sorted.bam GCF_000005845.2_ASM584v2_genomic.fna.fai (base) [b20223040323@admin1 test]$ cat GCF_000005845.2_ASM584v2_genomic.fna.fai ## 查看,第二列是每一条染色体的长度 chr1 4641652 6 80 81 chr2 39862 4699685 80 81 chr3 398 4740052 80 81
可见chr1的长度为:4641652。。。。。
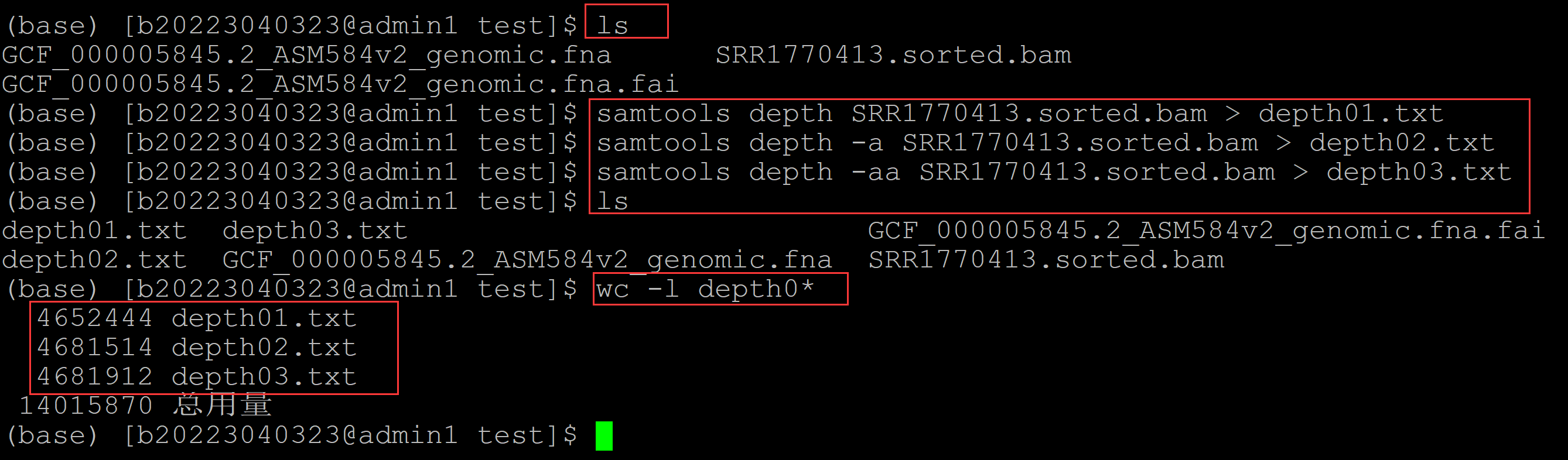
002、利用samtools的depth、depth -a、depth -aa分别对同一个bam文件进行测序深度统计 (base) [b20223040323@admin1 test]$ ls GCF_000005845.2_ASM584v2_genomic.fna SRR1770413.sorted.bam GCF_000005845.2_ASM584v2_genomic.fna.fai (base) [b20223040323@admin1 test]$ samtools depth SRR1770413.sorted.bam > depth01.txt ## depth (base) [b20223040323@admin1 test]$ samtools depth -a SRR1770413.sorted.bam > depth02.txt ## depth -a (base) [b20223040323@admin1 test]$ samtools depth -aa SRR1770413.sorted.bam > depth03.txt ## depth -aa (base) [b20223040323@admin1 test]$ ls depth01.txt depth03.txt GCF_000005845.2_ASM584v2_genomic.fna.fai depth02.txt GCF_000005845.2_ASM584v2_genomic.fna SRR1770413.sorted.bam (base) [b20223040323@admin1 test]$ wc -l depth0* ## 统计生成的depth文件的行数,可见三个文件行数不一致 4652444 depth01.txt 4681514 depth02.txt 4681912 depth03.txt 14015870 总用量
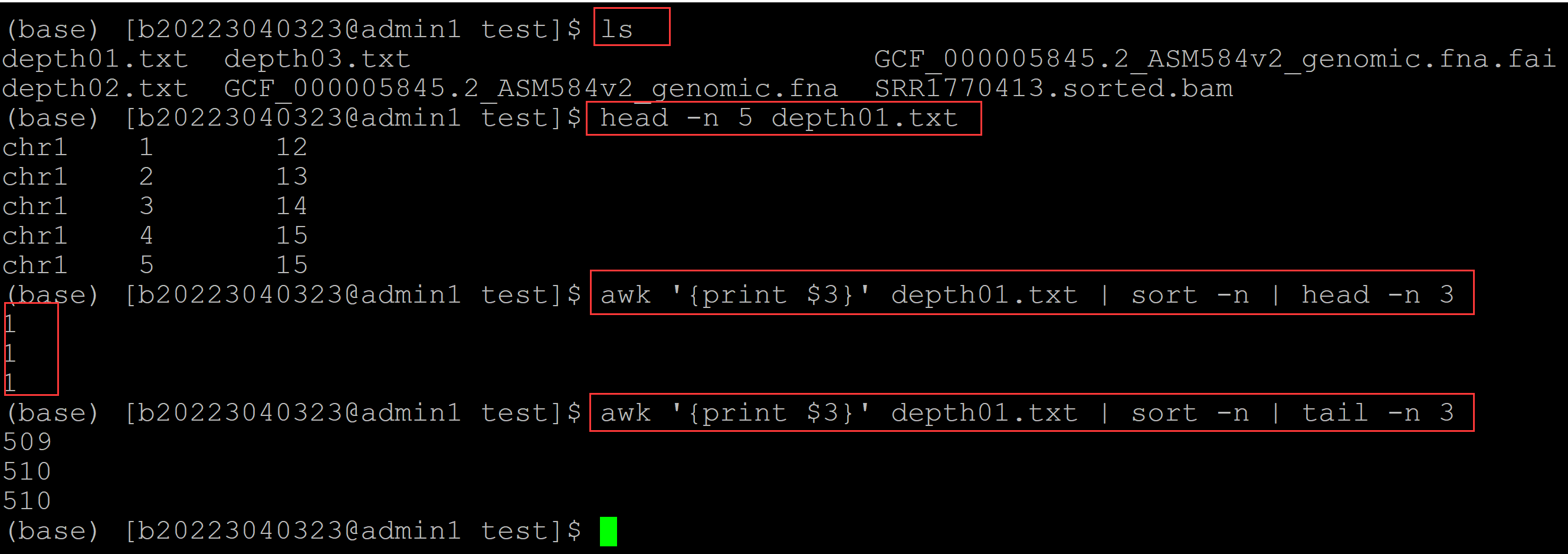
003、samtools的depth参数统计的是比对到参考基因组的染色体上所有位点测序深度大于0的所有测序深度数据,用一下脚本简单验证: (base) [b20223040323@admin1 test]$ ls depth01.txt depth03.txt GCF_000005845.2_ASM584v2_genomic.fna.fai depth02.txt GCF_000005845.2_ASM584v2_genomic.fna SRR1770413.sorted.bam (base) [b20223040323@admin1 test]$ head -n 5 depth01.txt ## depth01.txt文件记录的是位点的测序深度,每行一个位点 chr1 1 12 chr1 2 13 chr1 3 14 chr1 4 15 chr1 5 15 (base) [b20223040323@admin1 test]$ awk '{print $3}' depth01.txt | sort -n | head -n 3 ## 对第三列深度信息进行排序,可见最小深度为1 1 1 1 (base) [b20223040323@admin1 test]$ awk '{print $3}' depth01.txt | sort -n | tail -n 3 ## 输出最大测序深度 509 510 510
可见 samtools depth参数输出测序深度的结果最小的测序深度为1.
004、samtools depth -a参数输出的结果为fastq比对到参考基因组的染色体上的所有位点的测序深度,即在depth参数输出结果的基础上,也输出测序深度为0的位点。可用如下脚本进行验证: a、如果输出fastq数据比对到参考基因组的染色体上所有位点的测序深度, 那么depth02.txt的行数应该等于比对到染色体长度的总和,即chr1和chr2的总长度。 (base) [b20223040323@admin1 test]$ ls depth01.txt depth03.txt GCF_000005845.2_ASM584v2_genomic.fna.fai depth02.txt GCF_000005845.2_ASM584v2_genomic.fna SRR1770413.sorted.bam (base) [b20223040323@admin1 test]$ wc -l depth02.txt 4681514 depth02.txt (base) [b20223040323@admin1 test]$ cat GCF_000005845.2_ASM584v2_genomic.fna.fai chr1 4641652 6 80 81 chr2 39862 4699685 80 81 chr3 398 4740052 80 81 (base) [b20223040323@admin1 test]$ awk 'NR |
【本文地址】
今日新闻 |
推荐新闻 |