皮下脂膜炎样T细胞淋巴瘤:16例患者的临床特征、治疗方法和预后 |
您所在的位置:网站首页 › phoenix研究淋巴瘤 › 皮下脂膜炎样T细胞淋巴瘤:16例患者的临床特征、治疗方法和预后 |
皮下脂膜炎样T细胞淋巴瘤:16例患者的临床特征、治疗方法和预后
|
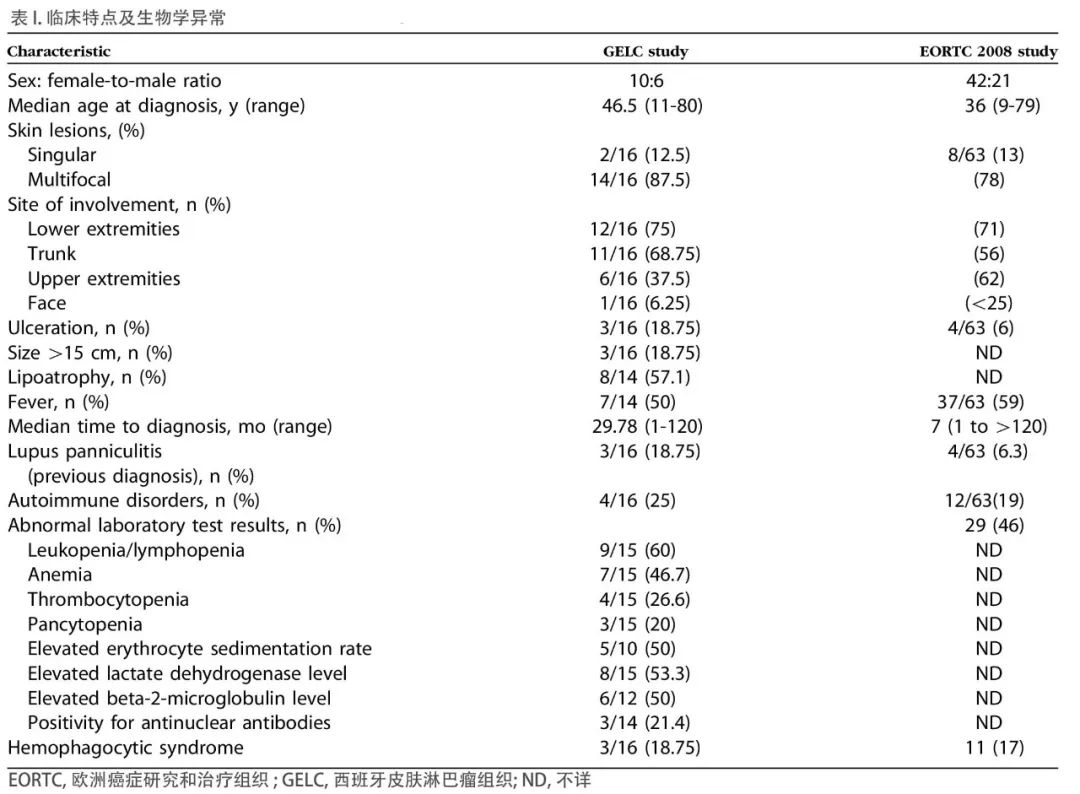
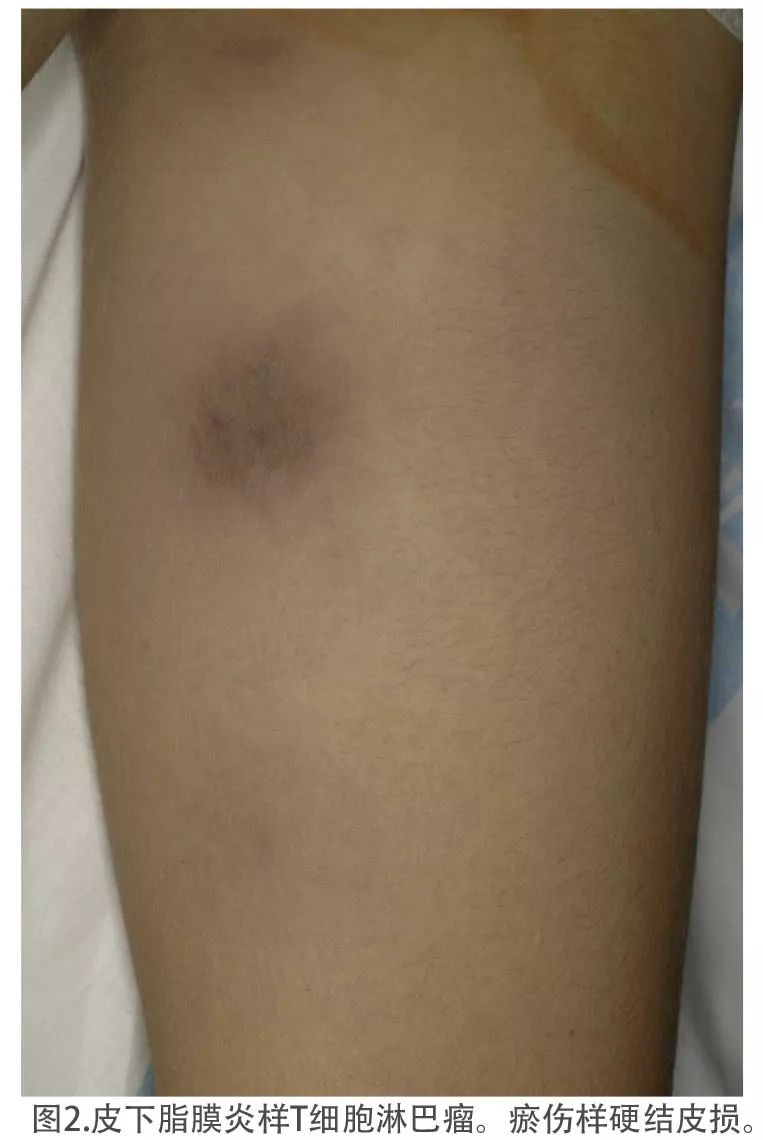
PET/CT:正电子发射断层显像 SPTCL: 皮下脂膜炎样T细胞淋巴瘤 内容提要 ● 皮下脂膜炎样T细胞淋巴瘤是一种罕见的原发性皮肤淋巴瘤。 ● 皮下脂膜炎样T细胞淋巴瘤不像以前认为的那样具有侵袭性,并且预后良好。 ● 免疫抑制疗法是一线治疗。 皮下脂膜炎样T细胞淋巴瘤(SPTCL)是一种罕见的成熟细胞毒性T细胞原发性皮肤淋巴瘤。在历史上,Gonzalez等人在1991年首次报道了这种情况,作为一种新型的T细胞淋巴瘤,它模拟了脂膜炎的临床症状。常常合并有噬血细胞综合征(HPS)(也称为噬血细胞淋巴组织细胞增生症)[1],随后被命名为SPTCL。最初,观察到该类型的淋巴瘤有了侵袭性的临床特征,因此建议进行积极的的多药综合化疗。然而,进一步的研究显示,根据T细胞表型(αβ或γδ),SPTCL在临床、组织学和免疫表型上存在差异,每种SPTCL的预后不同。2005年,世界卫生组织欧洲癌症研究和治疗组织(EORTC)皮肤淋巴瘤分类将SPTCL的定义限制为αβSPTCL。[2]在最近的分类中,SPLCT和原发性皮肤γδT细胞淋巴瘤被认为是两个明确而不同的疾病。 2008年,Willemze等人报告了EORTC皮肤淋巴瘤组的一个研讨会的结果,其中比较了63个αβSPTCL和20个γδSPTCL的数据。在那个时候,在大多数中心,治疗SPTCLs采用阿霉素为基础的综合化疗。结果表明,无HPS的αβSPTCL预后良好,以往作为首选治疗方案的综合化疗应受到质疑。[3] SPTCL对医生而言是一个挑战,因为除了诊断上的困难之外,目前还没有标准化的治疗方法。我们对16例SPTCL患者的临床资料进行回顾性分析。本研究的目的是:(1)更精确地确定临床特征和对治疗的反应,(2)将我们的发现与其他人群中先前的病例系列进行比较,(3)根据目前的知识状态找出哪些病例应该采用积极的治疗方法。 方法 在西班牙皮肤淋巴瘤组织的框架内,对1996年至2016年间确诊的16例SPTCL患者进行了回顾性多中心研究。资料取自9个皮肤淋巴瘤专业参考中心的皮肤科和病理科。获得了伦理审查委员会的批准。我们回顾所有与病人有关的临床信息,包括最初的表现和诊断历史、体格检查的描述、组织病理学、分期评估、治疗和临床过程。HPS是根据噬血细胞淋巴组织细胞增多症-2004标准定义的。[4]治疗反应的判断是基于国际工作组治疗反应标准。[5]随访时间从病理诊断日期到死亡日期或最后随访日期计算。 结果 患者人口统计数据和临床特征总结在表I中。临床上,患者仅表现为多发性结节和/或不同直径(通常小于10cm)的深度浸润斑块。皮损可有瘀伤样外观或表面轻微鳞屑的暗红色至粉红色的红斑(图1和2),在消退后它们可能遗留脂肪萎缩(图1)。有3例患者合并HPS(18.7%),无一例死亡。4例患者(25%)在诊断SPTCL之前患有相关的自身免疫疾病(即皮肤型红斑狼疮(2例)、多发性硬化和甲状腺功能减退)。
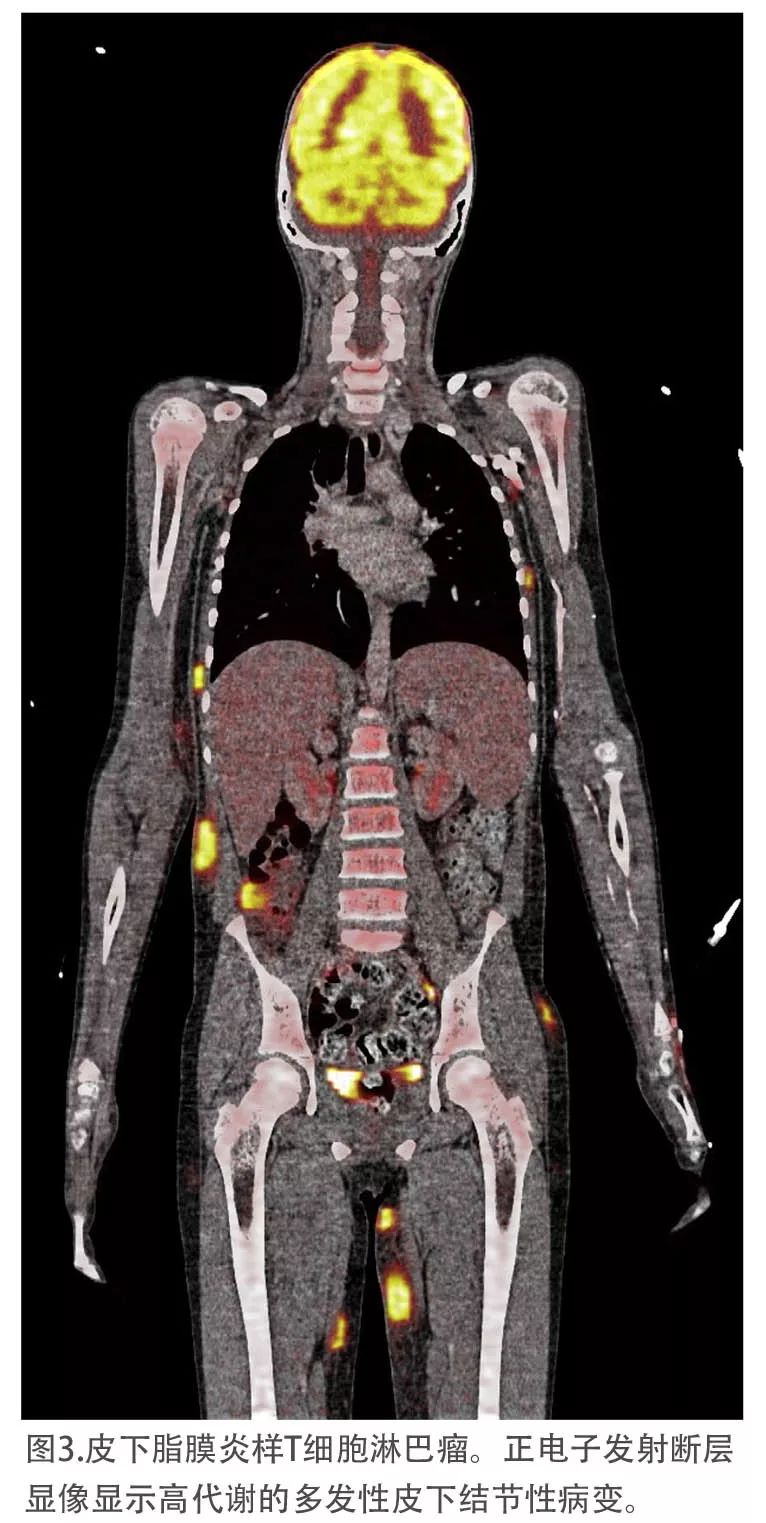
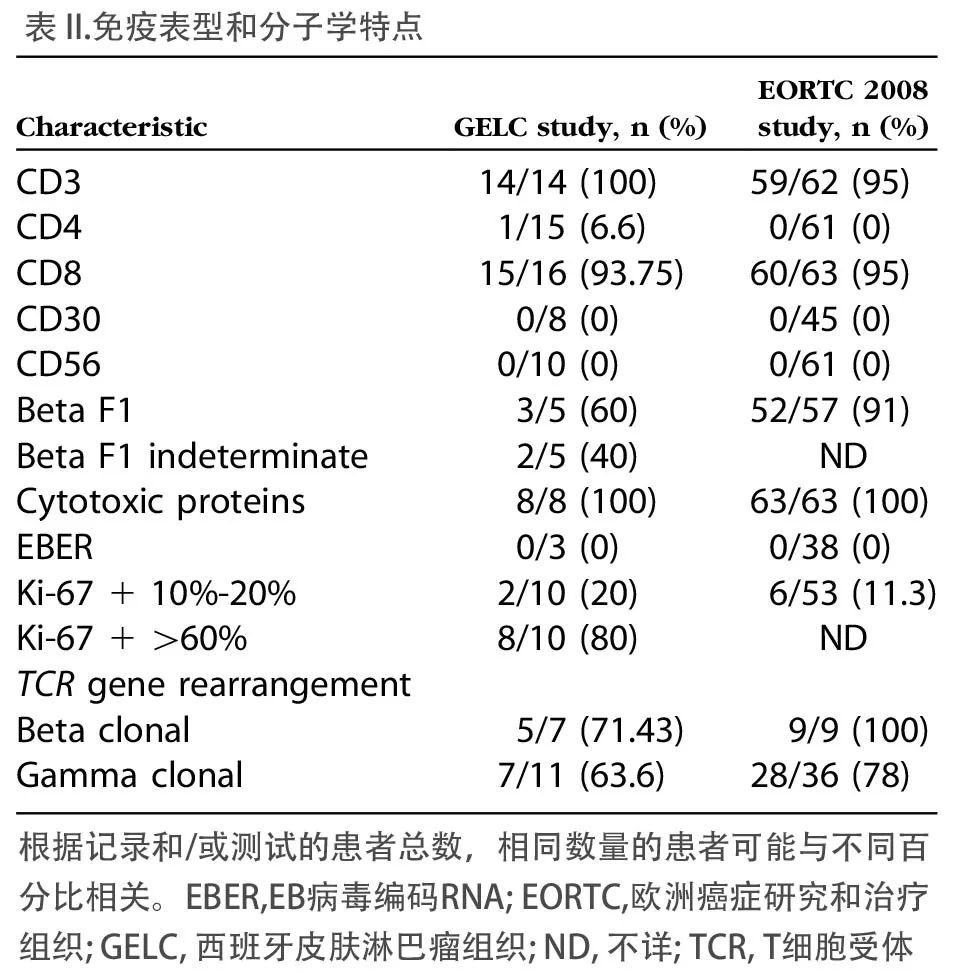
组织学上,所有用于诊断的皮肤活检标本均显示密集的皮下浸润,呈小叶性脂膜炎样改变。在细胞学上,非典型淋巴细胞通常是小到中等大小,具有不规则的深染核,并与小淋巴细胞、组织细胞和稀少浆细胞混合。肿瘤性T淋巴细胞在单个脂肪细胞(边缘化)周围浸润,通常显示CD3+、CD4-、CD8+、CD56-、TIA1细胞毒性颗粒相关RNA结合蛋白1阳性表型,具有高增殖率(表II)。影像学检查显示脾肿大(1例)和淋巴结肿大(2例)。3例患者均获得氟脱氧葡萄糖F 18(FDG)正电子发射断层显像(PET/CT)研究结果。皮损最大标准化摄取值为6.4~7.1。在这些患者中有1例患者有淋巴结受累(图3)。组织病理学上,淋巴结呈窦组织细胞增生,无噬血或淋巴瘤的迹象,但淋巴结周围的脂肪组织被不典型淋巴细胞浸润。在所有患者中,骨髓检查没有显示淋巴瘤的证据。1例患者出现噬血细胞增多症,另一例巨噬细胞数量增加。
单独口服类固醇或联合甲氨蝶呤或环孢素A是最常见的初始治疗(表III6030个月)发现2例HPS患者完全缓解。1例化疗后进行同种异体造血干细胞移植。平均随访时间35个月(中位随访时间145年生存率为85.7%。
讨论 SPTCL是一种少见的疾病,占所有皮肤T细胞淋巴瘤的1%以下。3本研究结果证实,SPTCL以女性为主,儿童和成人均受累。SPTCL最常见于50岁以下患者(3例中2例)。他们的中位诊断年龄为46.5岁,比其他病例系列稍大(这可能是由于我们的一些患者在诊断他们的SPTCL之前有长时间的皮损反复生长、消退的历史,以及包括了一个独特的小于20岁的患者)。这些发现不同于其他研究,其中诊断时的中位年龄为31-36岁[3,6,7],大约20%的患者年龄在20岁或更年轻。[3,7] 本研究的患者通常表现为多个无痛的皮下结节或硬化斑块,直径不同,主要累及下肢和/或躯干。一些病灶出现溃疡(18%的患者出现溃疡,高于此前报道的6%)。[3] 25%的患者有自身免疫性疾病史。与以往研究一致,最常见的自身免疫疾病是红斑狼疮,21%的研究病例检测到自身抗体。EORTC的研究报告了19%的病例与自身免疫性疾病有关。[3]法国最近的一项关于SPTCL的研究显示了最高的女性与男性比例(4.4),其中40%的患者曾被诊断出患有自身免疫疾病,65%的患者(其中一半患者没有自身免疫性疾病的病史)检测出自身抗体。[7] 在我们的研究中,18%的患者出现了HPS的发展,初诊时可以明确HPS的诊断,没有一例死亡。Willemze等报告了17%病例患有HPS;然而,63%的患者死亡。Michonneau等在他们的队列中发现37%的患者存在HPS,但未能将HPS作为生存的预后因素。[7]文献回顾16例日本发表的报告显示,B症状(81%的患者)和HPS(45%的患者)的发生率较高,而自身免疫性疾病的发生率较低(13%的患者)。[6] 本研究中SPLTCL的病理组织学表现与文献报道并无差异。值得注意的是,单靠多克隆T细胞受体(TCR)基因重排并不能排除SPTCL的可能性,尤其是只研究T细胞受体-γ链基因(TRG)时。我们能够在63%的患者中检测到T细胞受体重组的克隆,这低于EORTC研究报告的百分比。这种淋巴瘤的特殊形式可能影响这些结果。 SPTCL的诊断具有挑战性。红斑狼疮的临床特征可能与SPTCL的临床特征同时存在,即可能存在明显的重叠。[8-11] 氟脱氧葡萄糖F18的PET/CT正越来越多地被使用,在评估SPTCL的疾病程度和对治疗的反应方面是有用的。[12]非常重要的是,SPTCL罕有皮下组织外有淋巴瘤的证据,HPS可表现为肝脾肿大和淋巴结肿大。值得注意的是,在本研究中发现一例有淋巴结摄取FDG的患者,但随后的组织病理学检查并没有发现淋巴瘤的迹象,然而,SPTCL浸润了淋巴结周围的脂肪组织。因此,当观察到皮外摄取FDG的现象时,建议活检确认。近年来,已经描述了腹内脂肪参与SPTCL(肠系膜浸润伴中线腹部结节、降结肠系膜和胃肠系膜区)的情况。[13-15]PET/CT在检测腹腔内脏脂肪的隐匿性侵袭方面可能特别有用。[14,15]然而,其预后价值尚不清楚。 最常见的有文献记载的骨髓异常是噬血细胞综合征。[3]很少有脂肪细胞周围淋巴细胞分散灶累及骨髓形态的描述。在所有这类患者中,骨髓流式细胞术分析结果都不具有显著意义。[16,17]这一发现的发生率、意义及预后价值需要进一步评估。 由于SPTCL还没有建立标准的治疗方法,我们的患者接受了多种治疗。对于单发或局部皮损,建议放疗。[3]本研究中有2例患者使用局部放疗后获得成功,然而,在随后的病程中又出现新的皮损,后接受免疫抑制治疗。 这一结果和其他研究报告均表明,SPTCL患者对和以前接受过化疗的患者对免疫抑制药物的治疗有完全的反应。[7,18-16] 无HPS证据的患者可使用系统性糖皮质激素治疗或接受糖皮质激素与环孢霉素A及低剂量甲氨蝶呤等免疫抑制剂联合治疗,从而得到长期控制。在这些患者中,多药物化疗不应再被视为一线治疗。在大多数病例中,对环孢霉素A治疗的反应时间在2周内,仅用糖皮质激素治疗的反应时间在4周内。[19] 当药量逐渐减少或治疗停止时,复发是常见的,这不应被视为对治疗的抵抗。Guenova等人指出,维持缓解可能需要持续使用低剂量的糖皮质激素。[19] 伴有HPS的SPTCL患者通常不能单独使用传统的CHOP(环磷酰胺、阿霉素、长春新碱和强的松)化疗。对于合并HPS的患者,(大剂量)糖皮质激素联合环孢素A或造血干细胞移植支持的大剂量化疗可作为治疗选择。还需要进一步的合作研究,以确定SPTCL最佳的治疗策略。 https://doi.org/10.1016/j.jaad.2018.05.1243 本文原创,欢迎个人转发分享,其他任何媒体、网站如需转载或引用,须获得授权且在醒目位置注明出处。返回搜狐,查看更多 |


【本文地址】
今日新闻 |
推荐新闻 |