电子材料与器件计算方法基础 |
您所在的位置:网站首页 › 验算方法 › 电子材料与器件计算方法基础 |
电子材料与器件计算方法基础
|
问题
1
问题如下: 首先进入到需要的目录下,可以看到下列的文件列表 可以看到其中内容如图:
使用的是6-31G*基组 查阅相关资料,可以得到以下信息: 6-31G* 基组包含6个一般性质的基函数(6-31G),加上3个极化函数(d函数),共计9个基函数。其中,6个一般性质的基函数包括一个s型函数和三个p型函数,分别对应每个原子轨道的s和p轨道。 6-31G* 基组中的基函数可以用高斯型函数表示,即每个基函数都可以表示为多个高斯函数之和。这种表示方法使得计算效率更高,同时也更加准确,可以更好地描述分子的电子结构。 6-31G基组还包含了3个极化函数,使得该基组可以更好地描述分子中的电子云极化现象,从而更加准确地描述分子的电子结构。 在6-31G 基组中,每个一般性质的基函数(s和p轨道)都用三个高斯函数表示,而每个极化函数(d轨道)都用五个高斯函数表示。因此,6-31G基组共含有33个高斯函数(6个一般性质的基函数和3个极化函数,每个基函数用3或5个高斯函数表示)。 (b) i阅读上机说明,本人为第二组,需要修改run_job.sh 在文件夹目录下,使用bash执行文件 bash run_job.sh在输出文件1.out中进行搜索关于对称性的关键词 /symmetry输入?和/完成对上一条和下一条关键词的搜索,得到如下结果 D6H symmetry detected可以看到输入几何构型具有D6H对称性 查看优化后的原子分布: Output coordinates in angstroms (scale by 1.889725989 to convert to a.u.) No. Tag Charge X Y Z ---- ---------------- ---------- -------------- -------------- -------------- 1 C 6.0000 1.07904495 1.07904495 0.00000000 2 C 6.0000 -1.47400281 0.39495786 0.00000000 3 C 6.0000 0.39495786 -1.47400281 0.00000000 4 C 6.0000 -1.07904495 -1.07904495 0.00000000 5 C 6.0000 1.47400281 -0.39495786 0.00000000 6 C 6.0000 -0.39495786 1.47400281 0.00000000 7 H 1.0000 1.80736493 1.80736493 0.00000000 8 H 1.0000 -2.46890641 0.66154148 0.00000000 9 H 1.0000 0.66154148 -2.46890641 0.00000000 10 H 1.0000 -1.80736493 -1.80736493 0.00000000 11 H 1.0000 2.46890641 -0.66154148 0.00000000 12 H 1.0000 -0.66154148 2.46890641 0.00000000在优化后的苯分子中,最近邻C原子之间的距离是3.3986 Å。这是第1个C原子和第4个C原子之间的距离。 最近邻的C原子和H原子之间的距离是1.0857 Å。这是第1个C原子和第7个H原子之间的距离。 (b) ii搜索HOMO、LUMO,得到如下结果: HOMO = -0.248712 LUMO = -0.007899通过查阅资料得知 核磁共振(NMR)实验测得苯环相关参数为:HOMO = -7.2 eV, LUMO = -2.8 eV 紫外-可见(UV-Vis)光谱实验:HOMO = 6.5-7.5 eV, LUMO = 9.5-10.5 eV X射线光电子(XPS)谱实验:HOMO = 7.1-7.9 eV, LUMO = 10.6-11.3 eV 均与查阅得到的结果相去甚远,再回看题目,发现有提示如下: (提示: 输出中 LUMO 和HOMO 指示的数值可能不是最终的、计算后的 LUMO 和 HOMO 的值) 修改搜索关键词Molecular,得到以下结果: Vector 33 Occ=0.000000D+00 E= 5.013032D-01 Symmetry=e1u MO Center= 7.0D-15, 3.1D-15, -3.4D-34, r^2= 5.1D+00 Bfn. Coefficient Atom+Function Bfn. Coefficient Atom+Function ----- ------------ --------------- ----- ------------ --------------- 21 2.146089 2 C s 66 -2.146089 5 C s 37 1.977591 3 C px 82 1.977591 6 C px 7 1.393304 1 C px 52 1.393304 4 C px 6 -1.238745 1 C s 51 1.238745 4 C s 8 -1.181026 1 C py 53 -1.181026 4 C py可以看到HOMO能量为E= 5.013032D-01,比较来看,仿真与实际差异较大,可能一部分是因为参数为易于计算进行了一定程度的调整,同时仿真未完全的结果 结构优化步数可对照类似内容写: 结构优化步数:10 每一步循环次数 第一步:35 第二步:35 第三步:35 第四步:35 第五步:33 第六步:33 第七步:33 第八步:33 第九步:33 第十步:33 (c)修改run_bash.sh文件如下: #!/bin/bash nohup /opt/nwchem/bin/LINUX64/nwchem Benzene_for_electron_density_plot.nw >& 2.out &修改Benzene_for_electron_density_plot.nw文件如下: #以下是为了将来画出电子云而作的计算. echo restart BenzeneHF memory total 200 mb permanent_dir /home/dft_user26/home_work3/problem_1/problem_1/nwchem scratch_dir /home/dft_user26/home_work3/problem_1/problem_1/tmp task scf energy dplot title homo limitxyz -4.0 4.0 60 -4.0 4.0 60 -4.0 4.0 60 spin total gaussian output Benzene.cube end task dplot输入命令运行文件 bash run_bash.sh查看Benzene_electron_density.pdf结果(文件较大,打开速度较慢,请耐心等待) |

 由于不知道哪个是需要查看的运行配置文件,使用vim文本编辑器阅读运行文件
由于不知道哪个是需要查看的运行配置文件,使用vim文本编辑器阅读运行文件 可以看到,运行的是Benzene.nw文件,并将结果写入1.out文件 使用vim打开Benzene.nw
可以看到,运行的是Benzene.nw文件,并将结果写入1.out文件 使用vim打开Benzene.nw 据此可以回答第一个问题:
据此可以回答第一个问题: 由于我不知道mpi地址,姑且不使用并行计算了,改成下面的形式
由于我不知道mpi地址,姑且不使用并行计算了,改成下面的形式 安装好Gabedit,页面如图所示:
安装好Gabedit,页面如图所示:  将上述电子云运行结果文件Benzene.cube下载下来,放在不带有中文路径的位置上
将上述电子云运行结果文件Benzene.cube下载下来,放在不带有中文路径的位置上 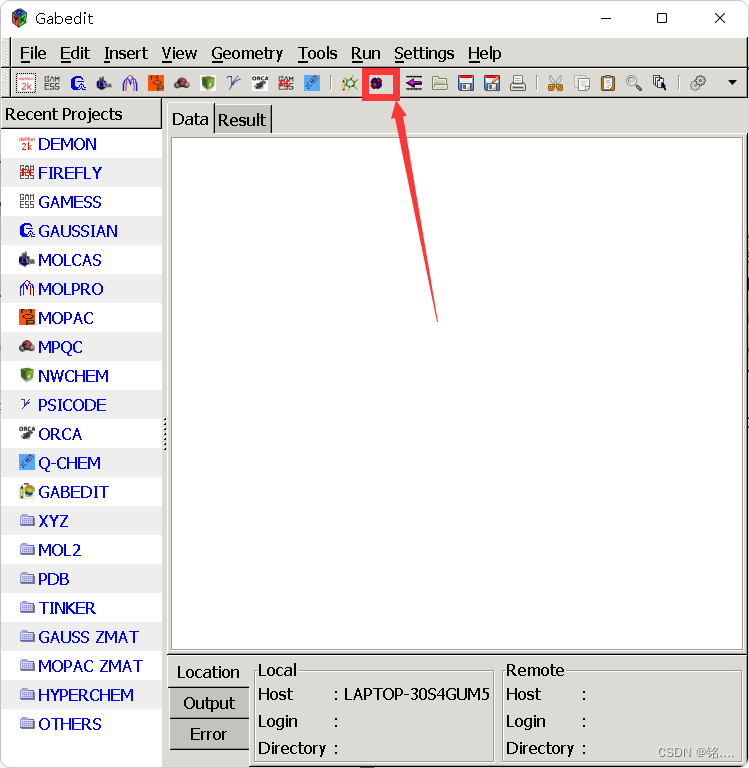 打开绘图
打开绘图 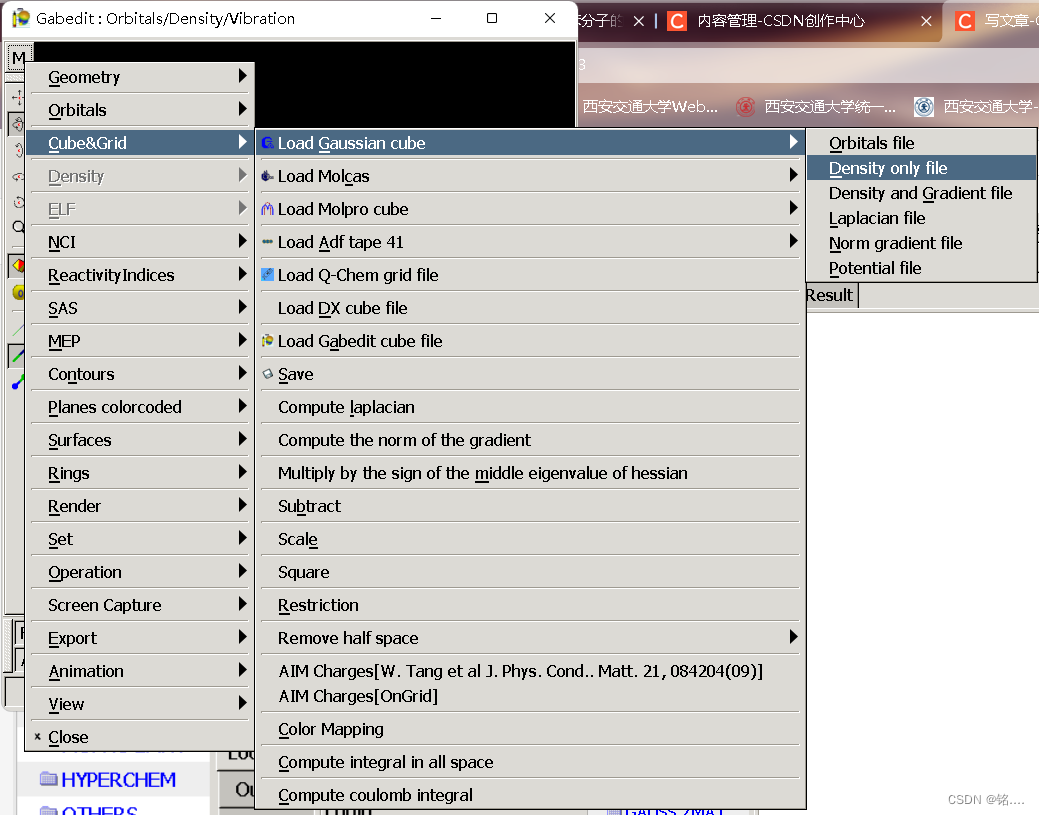 将cube文件导入,点击确定,结果如图
将cube文件导入,点击确定,结果如图 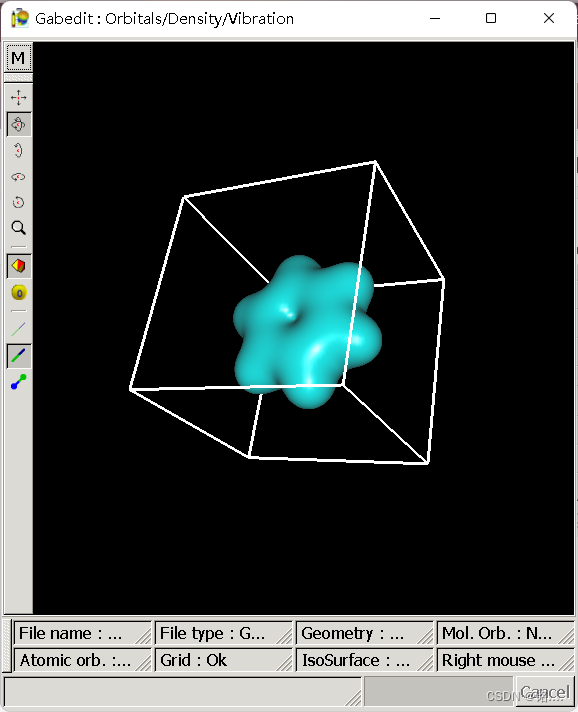 至此,第一问结束
至此,第一问结束【本文地址】
今日新闻 |
推荐新闻 |