Science经典:植物基因组的同线性与共线性分析思路 |
您所在的位置:网站首页 › 被子植物的祖先 › Science经典:植物基因组的同线性与共线性分析思路 |
Science经典:植物基因组的同线性与共线性分析思路
|
随着时间的推移,真核生物的基因组在基因同线性(synteny)和共线性(collinearity)方面存在一定程度的差异。这些差异在不同生物类群之间表现出不同的特征。 基因同线性(synteny,是指不同物种中这些基因都位于同一个染色体区域,collection of contiguous genes located on the chromosome of different species) 共线性(collinearity,是指不同物种中这些基因都位于同一个染色体区域,且这些基因在染色体上的排列顺序相同, a particcular kind of synteny in which the genes are conserved in the same order)例如,对于大多数真兽目(胎盘哺乳动物)类群来说,从约1.3亿年前的共同祖先开始,染色体片段的重排程度相对较小。确实,从推测发生于约5亿年前的全基因组复制事件至今,主要脊椎动物谱系的染色体组成(核型)的演化一直比较缓慢。因此,在真兽目类群中,准确识别同源基因通常相对容易,而根据“基因组中的最佳匹配标准” (best-in-genome) 进行共线性和同线性的推断通常也相对简单。这一标准是通过在两个基因组之间进行配对比较,识别出一对一最佳匹配的染色体区域。 然而,在被子植物(开花植物)中,情况有所不同。被子植物的基因组在大小和结构方面表现出显著的波动,即使在亲缘关系较近的种群之间也存在很大差异。在过去的约2亿年中,被子植物经历了多次全基因组复制事件,伴随着大规模基因丢失,导致祖先基因分散在多条染色体上。被子植物的基因组大小超过了原来的1000倍,其中一部分差异主要存在于异染色质区域。此外,由于移动元件引起了短DNA片段的大规模重排,导致异染色质区域的大规模共线性几乎完全消失。 尽管被子植物经历了多次全基因组复制,但染色体数量比基因组大小稳定,大多数类群的染色体数目在小于50倍的范围内波动。染色体融合是一个常见的现象,发生在许多生物谱系中,其中一个典型的案例是高粱属植物,其中染色体数为 10 (n = 10)的植物是与染色体数为 5(n = 5)的植物的祖先。玉米和高粱尽管拥有相同的染色体数(n = 10),但它们分别经历了不同的基因组复制事件,可能是由多次染色体融合事件导致的。这也解释了为什么玉米10号染色体和5号染色体的单臂与整个高粱6号染色体和4号染色体相对应。 全基因组测序的应用使我们能够使用统一的理论框架来分析不同物种的进化关系,尤其是染色体的对应关系。在被子植物中,同线性和古多倍化分析密切相关,因为被子植物的比较基因组学分析必须考虑到基因组复制和分散分布的影响。以拟南芥为例,它经历了三次古多倍化事件(2次二倍化和1次三倍化),导致祖先染色体组产生了12个拷贝分散于大约160 Mb碱基的基因组中。在过去的几百万年中,拟南芥与其他植物如岩生拟南芥和粉红草袋进化为不同分支时,发生了8-9次染色体重排事件,包括6 条染色体融合成 3 条染色体导致染色体数目从 8 变为 5,这使得拟南芥与其他被子植物进行比较基因组学分析变得异常复杂。 相比之下,其他双子叶植物的基因组结构相对较简单。杨树经历了特定于其杨柳科植物谱系的一次基因组复制事件,与拟南芥共享古多倍化事件中的一次(γ)。葡萄和番木瓜每个物种只经历了γ事件,没有发生后续的多倍化事件。特别值得注意的是,番木瓜(从物种分类上与拟南芥同属十字花目)中不存在β事件,而对葡萄基因组的分析认为β事件发生在拟南芥-杨树的共同祖先中,因此两者存在冲突。 同线性(synteny)可通过相邻匹配的基因对的成簇来鉴定,但在不同物种之间,由于基因密度和串联基因簇的差异,可能会导致统计误差。共线性(collinearity)则是同线性的一种更具体的表现形式,要求基因对在不同物种中具有相同的排列顺序。通常,同线性和共线性是通过查找一对一的保守性来确定的,这意味着两个物种之间的对应基因在演化中保持不变。为了更好地利用新的基因组数据资源,需要进行多向共线性分析,包括渐进比对 (progressive alignments),伴随统计评估和迭代优化。在被子植物中,这种多向比对的方法有助于揭示基因组复制带来的影响。 在被子植物中,推断祖先基因顺序的一种方法是自下而上的方法,其中最近复制的片段被交错合并生成假设的中间体,然后逐步合并。然而,这种方法需要为每个复制事件进行额外的推断循环,并且可能会积累错误。另一种自上而下的替代方法只需要进行一次推断循环,同时搜索并对齐多个基因组和亚基因组(subgenome)中的所有结构相似的片段。自上而下的方法通常更灵敏,因为它可以包括传递同源性 (transitive homology, 或称为间接同源性),即片段A和B可能经历了相对应的同源基因丢失,不再直接对应,但两者仍与片段C对应。通过与不具有复制事件或独立基因丢失的另一个基因组进行比较,可以解决自下而上的方法可能忽略的复制区域退化的关系。利用这种比较方法,研究人员揭示了酵母物种之间的同线性。 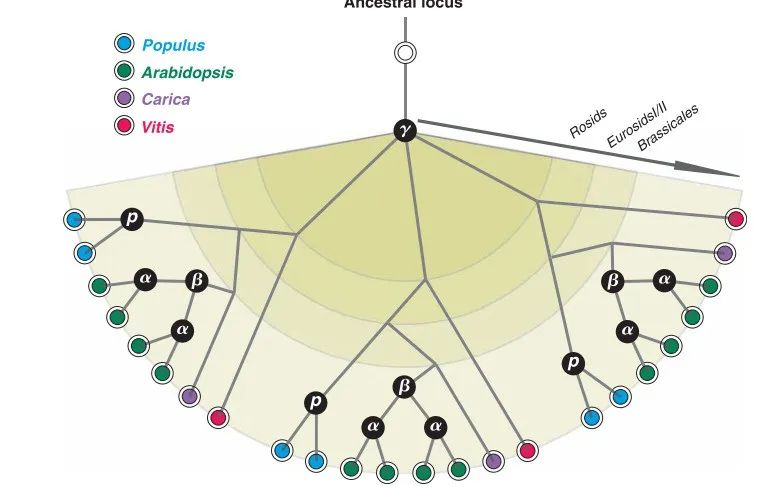 图1. 理想化的基因树,其中包含了在杨树、拟南芥、番木瓜和葡萄中的多个直系同源基因和旁系同源基因。为了说明问题,这里假设在所有分支上的进化速率相等,并且多倍体化后没有基因丢失。黑色圆圈代表多倍体化事件,α和β表示拟南芥(Arabidopsis )谱系的多倍体化,p表示杨树(Populus )的基因组复制事件,γ表示所有四个物种之间共享的多倍体化事件。 自上而下的分析结果显示,拟南芥(Arabidopsis)、番木瓜(Carica)和杨树(Populus)之间存在高度的共线性。例如我们鉴定出的包含多个同源片段的3个分支中,每个分支包括来自最多四个拟南芥、一个番木瓜和两个杨树基因组区域,这表明这些物种的共同祖先可能经历了古六倍体事件【这里没看明白,怎么能根据这个图就推断出是古六倍体事件?难道是前面提到的 2 个二倍体化、1 个 3 倍体化产生 12 个拷贝,对应这一个六倍体化产生 12 个拷贝?】。利用这些方法对葡萄基因组的分析验证了推断重建的基因排列顺序和拟南芥-番木瓜-杨树共同祖先的三重结构(图2A)。葡萄作为一个独立的外群可以用来测试基因顺序的比对,而古六倍体被认为覆盖了葡萄基因组的大部分。当拟南芥-番木瓜-杨树的同源序列比对到葡萄时,两个独立推断的三倍体化模式非常相似(图2B)。因此,自上而下的基因顺序比对揭示了在拟南芥和杨树中未曾被检测到的基因组三倍体化事件,并揭示了这一事件发生在葡萄、拟南芥、番木瓜和杨树的共同祖先中。  图2. 多个双子叶植物基因组共线性区域展示 三角形代表基因及其转录方向。没有与展示的区域同源的基因未绘制。(A)拟南芥(Arabidopsis ,绿色)、番木瓜(Carica , 品红色)和杨树(Populus, 蓝色)染色体区域之间的对比。比对结果揭示了四个不同的重复事件,如图1所示。这些区域根依据简约性原则分为三个一致的γ-亚基因组(Con γA、γB、γC)。每个γ-亚基因组中比对上的基因基于一致性原理合并成一个推断的顺序。(B)通过葡萄基因组(Vitis, 红色)验证了推测的γ分区,因为(A)中的每个γ-亚基因组仅有一个与葡萄染色体区域密切匹配。 随着更多基因组的测序完成,被子植物基因和基因组的比较进化分析将在其性能和精确性上有更大改进。然而,目前的研究框架仍然表现出两极性,因为在核心真双子叶植物和禾本科植物内,可以识别广泛的同线性和共线性,但因为更长的进化距离和更多的基因组重排,在他们之间的同线性和共线性较少。以水稻(Oryza sativa)为例,与四个核心真双子叶植物之间的共线同源基因仅占Oryza基因的大约15%,且仅分布在大约一半的基因组中。最长的水稻-拟南芥共线片段包含23对同源基因对,但加入葡萄(Vitis)基因组后,同源基因对的数量可以提高到47对。来自非谷物基因组的其他单子叶植物序列,如香蕉(Musa acuminata)或菠萝(Ananas comosus),以及来自基部真双子叶植物,如加利福尼亚罂粟或鸦片罂粟(Papaver somnifera),以及基部被子植物,如Amborella trichopoda(无通用名),可能会进一步增加跨不同被子植物类群的共线性和同线性检鉴定。 被子植物泛基因组比较揭示了古多倍体谱系中基因保留和丢失的相关模式。在多倍化事件后,多个后代染色体的比对显示了一些祖先基因能够抵抗缺失,它们一直被保留在同线性亚基因组中。这种对特定基因家族(如MADS-box基因)和其他转录因子的优先保留可能会增加形态复杂性。另一方面,在多次多倍化循环后,具有单一拷贝的基因功能组的成员始终只存在一个拷贝,这表明这些基因具有特定的优势。 考虑到植物之间DNA替代速率的可变性,偏离共线性可能是一种更可靠的系统发育特征。DNA替代速率在种子植物谱系中是高度可变的,甚至在同一属内的替代速率也可能相差100倍。因此,分析基因组结构中稀有变化(相对于DNA替代速率)的特点,如基因的特异性重排、插入或缺失,可能是分析许多谱系进化关系的信息丰富且稳健的方法。 自上而下的比对方法为从多个基因组和亚基因组中获得的共线性和同线性比对提供了很大的改进,这些比对可能成为重建被子植物基因组祖先状态的基础。通过自上而下的比对,可以估算出同线性区块内共有基因的顺序。在宏观水平上,同线性区块之间的顺序则难确定;然而,有几种组合算法可根据最简约的重排算法来重建祖先基因组。这样获得的顺序将不仅揭示共有的基因,还揭示了插入到新位置的不同基因,突显了谱系特异性的变化。更多基因组序列将促进在微观水平鉴定基因顺序,同时有助于鉴定基因组中重要的功能DNA片段,例如类似于28个脊椎动物基因组中在进化上受到限制的功能元件。 文献: H. Tang, J. E. Bowers, X. Wang, R. Ming, M. Alam and A. H. Paterson Science 2008 Vol. 320 Issue 5875 Pages 486-8 |
【本文地址】
今日新闻 |
推荐新闻 |