蛋白质二级、三级结构预测 |
您所在的位置:网站首页 › 蛋白质的序列比其空间结构更保守 › 蛋白质二级、三级结构预测 |
蛋白质二级、三级结构预测
|
一.蛋白质结构预测
1.什么是蛋白质结构预测?
蛋白质是生命活动的基本单位,其结构决定了功能,对蛋白质结构的研究有助于对其功能的研究。上世纪50年代初,Anfinsen等人提出蛋白质的空间结构是由其一级结构决定。 蛋白质都是由20种不同的L型α氨基酸连接形成的多聚体,在形成蛋白质后,这些氨基酸又被称为残基。 测定蛋白质序列比测定蛋白质结构容易得多,而蛋白质结构可以给出比序列多得多的关于其功能机制的信息。 蛋白质结构预测指的是:已知氨基酸序列,通过计算的手段预测蛋白质的二级结构和空间三维结构。 获得蛋白质 序列数据要比获得结构数据简单得多, DNA测序 技术的突飞猛进更使得可直接通过翻译、推导得 到大量的蛋白质序列. 而目前蛋白质结构数据库 PDB中所存储的蛋白质三维结构主要通过X 射线晶体衍射和核磁共振成像技术得到, 两种实验方法 均成本不菲, 且有各自的应用局限. 截止2016年 5 月, PDB 数据库中存储了11万余条蛋白质结构 数据, 而这只占UniProt中所有蛋白质序列数据的 1/600, 也就是说只有不到0.2%的蛋白质序列拥有 实验测定的三维结构。(来自2016年的物理学报文章) 2.蛋白质结果预测如何实现?
(2)无模板的结构预测方法——从头预测方法 从头预测方法不依赖于任何已知结构, 而是以第一性原理构建蛋白质折叠力场, 再通过相应的构象搜索方法搜寻目标蛋白的天然结构. 第一性原理是从头计算,不需要任何参数,只需要一些基本的物理常量,就可以得到体系基态的基本性质的原理。 无模板的结构预测方法与基于模板的预测方法的关键区别在于: 它们并不依赖于任何一个完整的结构模 板, 不要求片段所在模板与目标蛋白有任何同源性 或结构相似性, 这让它们具有更大的随机性和自由 度, 便于模拟已知结构中不存在的全新结构. 1. 构象初始化 “基于模板” 和“无模板” 两类结构预测方法的关键区别——就在于所采取的构象初始化方法不同: 基于模板的预测方法通过搜索识别与目标蛋白具有同源性或结构相似性的已知结构作为模板而获得初始化构象;(提高序列比对方法的效率、精度是促进同源建模方法发展的关键.) 无模板的预测方法则通常以小的结构片段为起点从头构建初始构 |
 蛋白质的分子结构可划分为四级,以描述其不同的方面: • 蛋白质一级结构:组成蛋白质多肽链的线性氨基酸序列。 • 蛋白质二级结构:依靠不同氨基酸之间的C=O和N-H基团间的氢键形成的稳定结构,主要为α螺旋和β折叠。 • 蛋白质三级结构:通过多个二级结构元素在三维空间的排列所形成的一个蛋白质分子的三维结构。 • 蛋白质四级结构:用于描述由不同多肽链(亚基)间相互作用形成具有功能的蛋白质复合物分子。
蛋白质的分子结构可划分为四级,以描述其不同的方面: • 蛋白质一级结构:组成蛋白质多肽链的线性氨基酸序列。 • 蛋白质二级结构:依靠不同氨基酸之间的C=O和N-H基团间的氢键形成的稳定结构,主要为α螺旋和β折叠。 • 蛋白质三级结构:通过多个二级结构元素在三维空间的排列所形成的一个蛋白质分子的三维结构。 • 蛋白质四级结构:用于描述由不同多肽链(亚基)间相互作用形成具有功能的蛋白质复合物分子。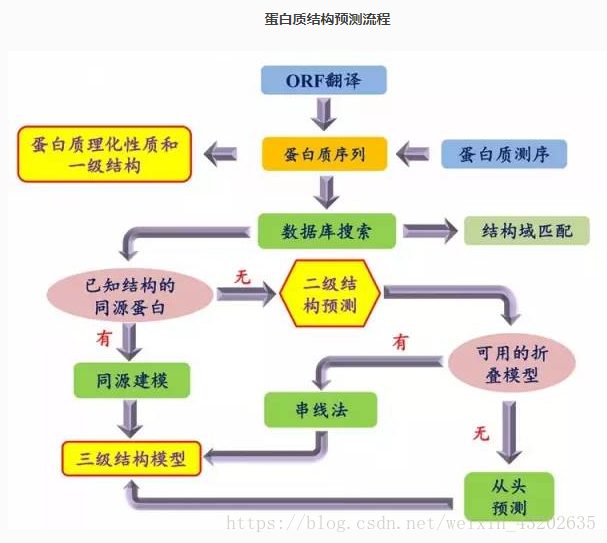 (1)基于模板的结构预测方法:同源建模+穿线方法 由于相似的蛋白质序列往往拥有相似的三维结构, 这就有了以PDB数据库中的已知结构为模板的同源建模方法, 它是迄今为止精度最高的一类结构预测方法.(来自2016年的物理学报文章) 而当PDB数据库中找不到与待预测蛋白质序列(下文中称为“目标 蛋白”或“目标序列”)具有显著序列相似性的蛋白质结构时, 此时通过穿线方法仍有可能找出与目标蛋白具有结构相似性的已知结构. 穿线方法——穿线方法实际上是通过某种策略将序列与结构进行比对, 评估将序列以各种匹配方式“安放”到各个三维结构上的“舒适”程度, 因此也被称为折叠辨识。 注:由于蛋白质结构 远比序列具有更高的保守性,毫无序列相似性的两个蛋白质也可能拥有相似的结构, 这是穿线法发挥作用的领域.
(1)基于模板的结构预测方法:同源建模+穿线方法 由于相似的蛋白质序列往往拥有相似的三维结构, 这就有了以PDB数据库中的已知结构为模板的同源建模方法, 它是迄今为止精度最高的一类结构预测方法.(来自2016年的物理学报文章) 而当PDB数据库中找不到与待预测蛋白质序列(下文中称为“目标 蛋白”或“目标序列”)具有显著序列相似性的蛋白质结构时, 此时通过穿线方法仍有可能找出与目标蛋白具有结构相似性的已知结构. 穿线方法——穿线方法实际上是通过某种策略将序列与结构进行比对, 评估将序列以各种匹配方式“安放”到各个三维结构上的“舒适”程度, 因此也被称为折叠辨识。 注:由于蛋白质结构 远比序列具有更高的保守性,毫无序列相似性的两个蛋白质也可能拥有相似的结构, 这是穿线法发挥作用的领域.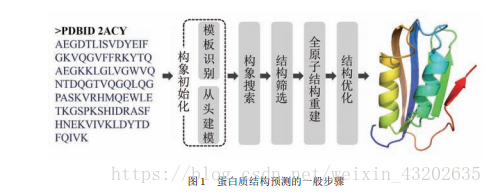
【本文地址】
今日新闻 |
推荐新闻 |