Plasmer:基于共享k |
您所在的位置:网站首页 › 细菌基因组的提取结果分析怎么写 › Plasmer:基于共享k |
Plasmer:基于共享k
|
Plasmer:基于共享k-mers和基因组特征利用机器学习算法的一个准确且敏感的细菌质粒预测工具 Plasmer: an Accurate and Sensitive Bacterial Plasmid Prediction Tool Based on Machine Learning of Shared k-mers and Genomic Features
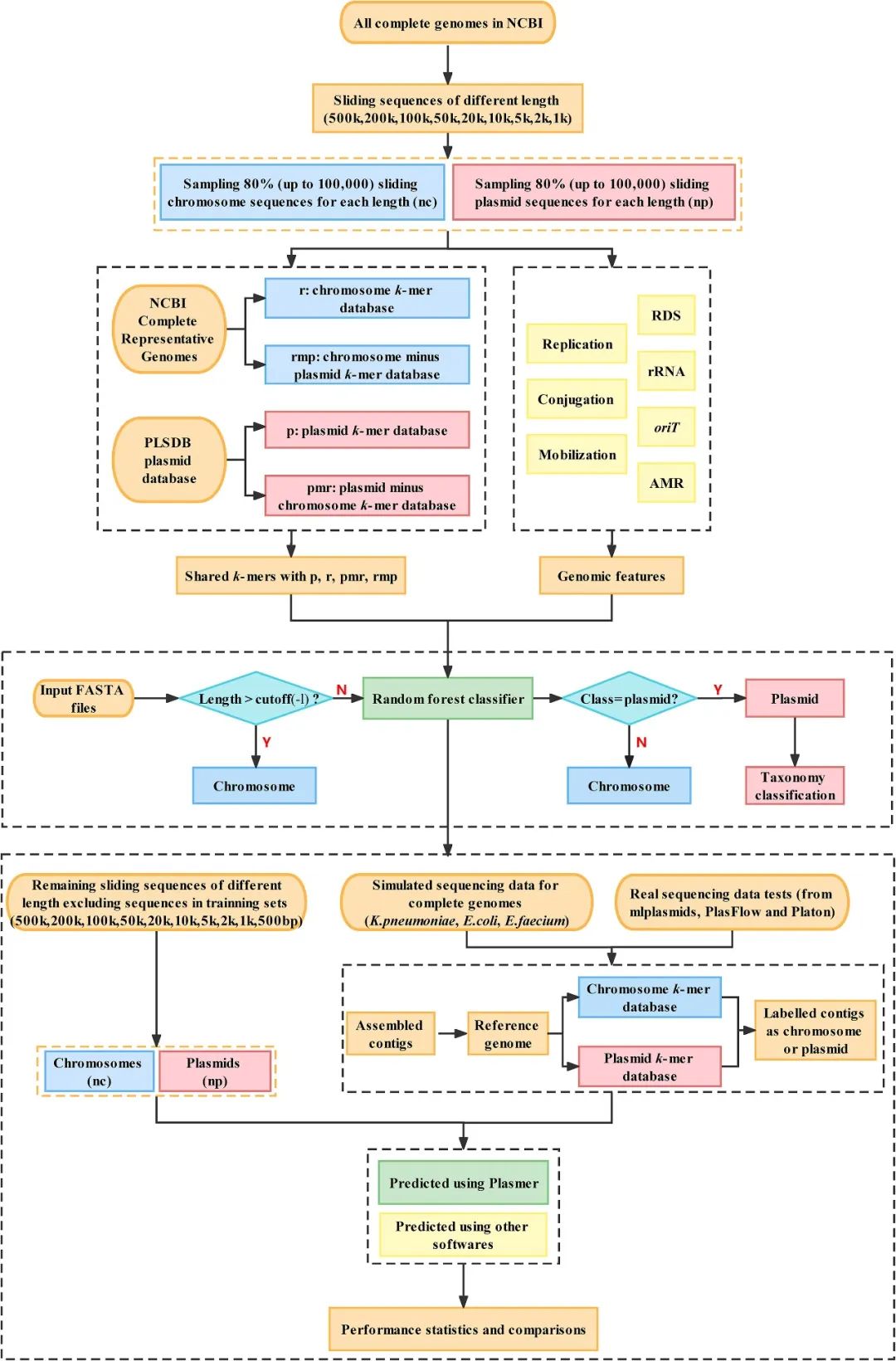
Microbiology Spectrum,[IF 9.043] DOI:10.1128/spectrum.04645-22 原文链接:https://journals.asm.org/doi/epub/10.1128/spectrum.04645-22 第一作者:Qianhui Zhu (朱倩慧);Shenghan Gao (高胜寒) 通讯作者:Songnian Hu (胡松年);Zilong He (贺子龙) 合作作者:Binghan Xiao (肖冰寒) 主要单位: 中国科学院微生物所 (State Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China) 中国科学院大学 (University of Chinese Academy of Sciences, Beijing, China) 北京航空航天大学医学科学与工程学院(School of Engineering Medicine, Beihang University, Beijing, China) 生物力学与力生物学教育部重点实验室(Key Laboratory of Biomechanics and Mechanobiology (Beihang University), Ministry of Education) 大数据精准医疗工信部重点实验室(Key Laboratory of Big Data-Based Precision Medicine (Beihang University), Ministry of Industry and Information Technology of the People’s Republic of China) - 摘要 - 细菌基因组中质粒的鉴定对许多因素至关重要,包括水平基因转移、抗生素抗性基因、宿主-微生物相互作用、克隆载体和工业生产。目前已有一些生物信息学方法来预测组装基因组中的质粒序列。然而,现有的方法有明显的缺点,如敏感性和特异性不平衡,依赖特定物种的模型,以及在短于10kb的序列中性能下降,限制了它们的适用范围。Plasmer是一种利用机器学习算法基于共享k-mers比例和基因组特征的新型质粒预测工具。与现有的基于k-mer频率或基因组特征的方法不同,Plasmer采用随机森林算法,利用序列与质粒和染色体k-mer数据库共享k-mer的百分比,结合基因组特征进行预测,包括比对E-value和复制子分布得分(replicon distribution scores, RDS)。Plasmer对所有物种均适用,其AUC为0.996,准确率为98.4%。与现有的方法相比,对滑窗切分序列和模拟测序组装contigs及真实测序数据de novo组装contigs的测试结果一致表明,Plasmer在500bp以上的长短片段上具有优于其他方法的准确性和稳定的性能,表明其对碎片化组装的适用性。对于长度大于500 bp的序列,Plasmer的灵敏度和特异性均大于0.95,消除了现有方法中常见的灵敏度或特异性的偏差,另外,在所有方法中,Plasmer 的F1 score最高。Plasmer还提供了质粒的分类学分类。 - 亮点 - 在这项研究中,我们提出了一个名为Plasmer的新型质粒预测工具。与现有的基于k-mer或基因组特征的方法不同,Plasmer是第一个将共享k-mers百分比和基因组特征结合起来的工具。这使得Plasmer性能有了明显的提高,(i)在滑窗切分序列和模拟测序组装contigs及真实测序数据de novo组装contigs上均具有最高的F1 score和准确性;(ii)适用于500bp以上的片段,具有最高的准确性,能够在碎片化的组装结果中进行质粒预测;(iii)在敏感性和特异性之间有出色和平衡的表现(500bp以上均>0. 95),消除了其他方法中普遍存在的灵敏度或特异性方面的偏差;(iv)不依赖特定物种训练模型。Plasmer为细菌基因组组装结果中的质粒预测提供了一个更可靠的选择。 - 结果 - 1. Plasmer工作流程概述 Overview of the Plasmer workflow Plasmer的工作流程(图1)包括三个阶段:(i)数据准备和k-mer数据库的构建;(ii)序列特征提取和机器学习模型构建,以及(iii)使用机器学习模型对输入序列进行预测。首先,Plasmer判断每个输入序列的长度,长于给定参数(-l 默认为500 kb)的序列被直接标记为染色体序列。第二,计算每个输入序列与质粒和染色体k-mer数据库的共享k-mers比例。第三,Plasmer整合了更高水平的基因组特征,如RDS、接合基因、oriT和rRNA,以提高整体性能。接下来,使用从NCBI完整细菌基因组的不同长度滑窗序列中分层抽取的约150万条序列训练随机森林分类器,并在剩余滑窗序列上进行了性能测试。最后,对来自模拟和真实测序数据的组装结果进行了性能测试。
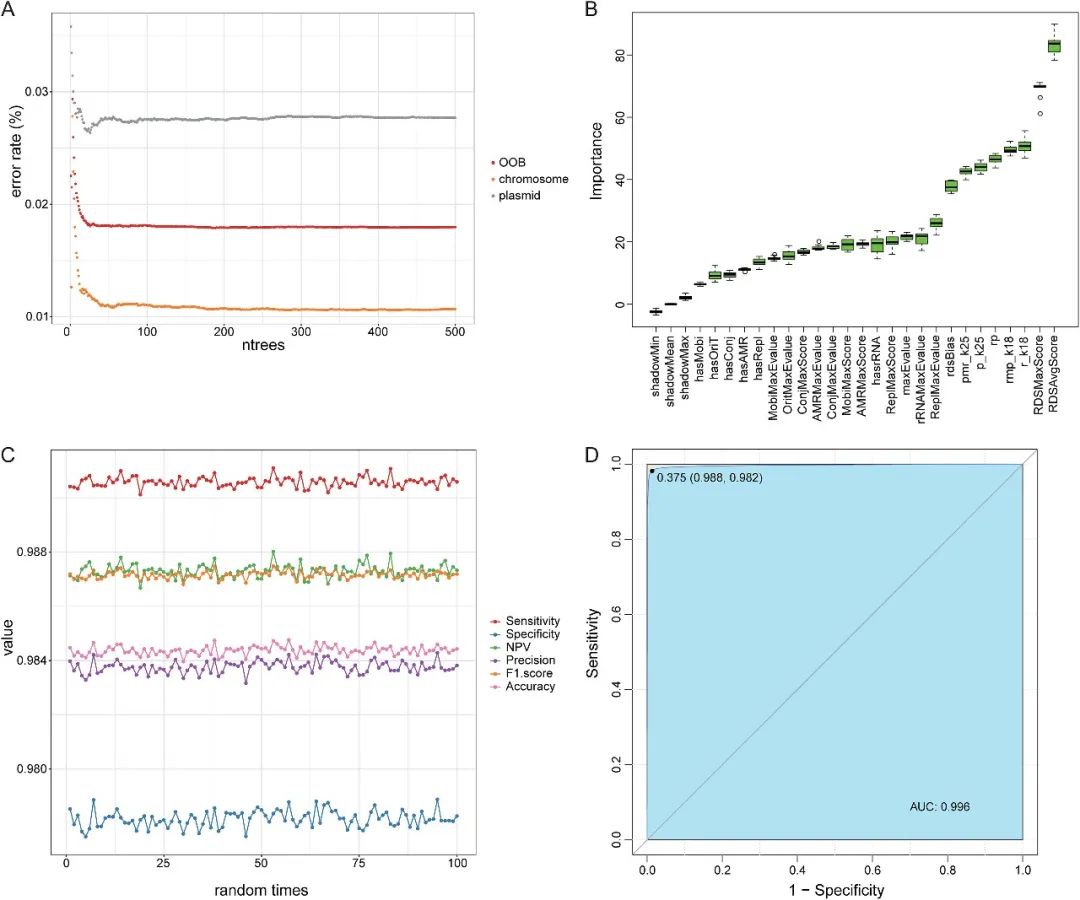
图1 Plasmer 流程图 2. 随机森林模型区分质粒和染色体序列性能出色 The random forest model had excellent performance in classifying plasmid and chromosomal sequences 在模型训练之前,我们评估了随机森林中决策树的数量和所有特征的重要性。当树的数量增长到200棵以上时,随机森林模型的误差逐渐收敛(图2A),证实了我们选择树的数量的可行性(ntrees = 500)。同时,最重要的特征是RDS和k-mer相关的特征。结果显示(图2B),所有的特征均是重要的,因此全部用于训练。经过100次训练集和测试集的随机抽样以及随机森林建模,模型准确率约为98.5%,灵敏度为0.99左右;F1 score和精确度等都在0.98以上,特异性也接近于0.98。(图2C)。模型的AUC为0.996(图2D)。
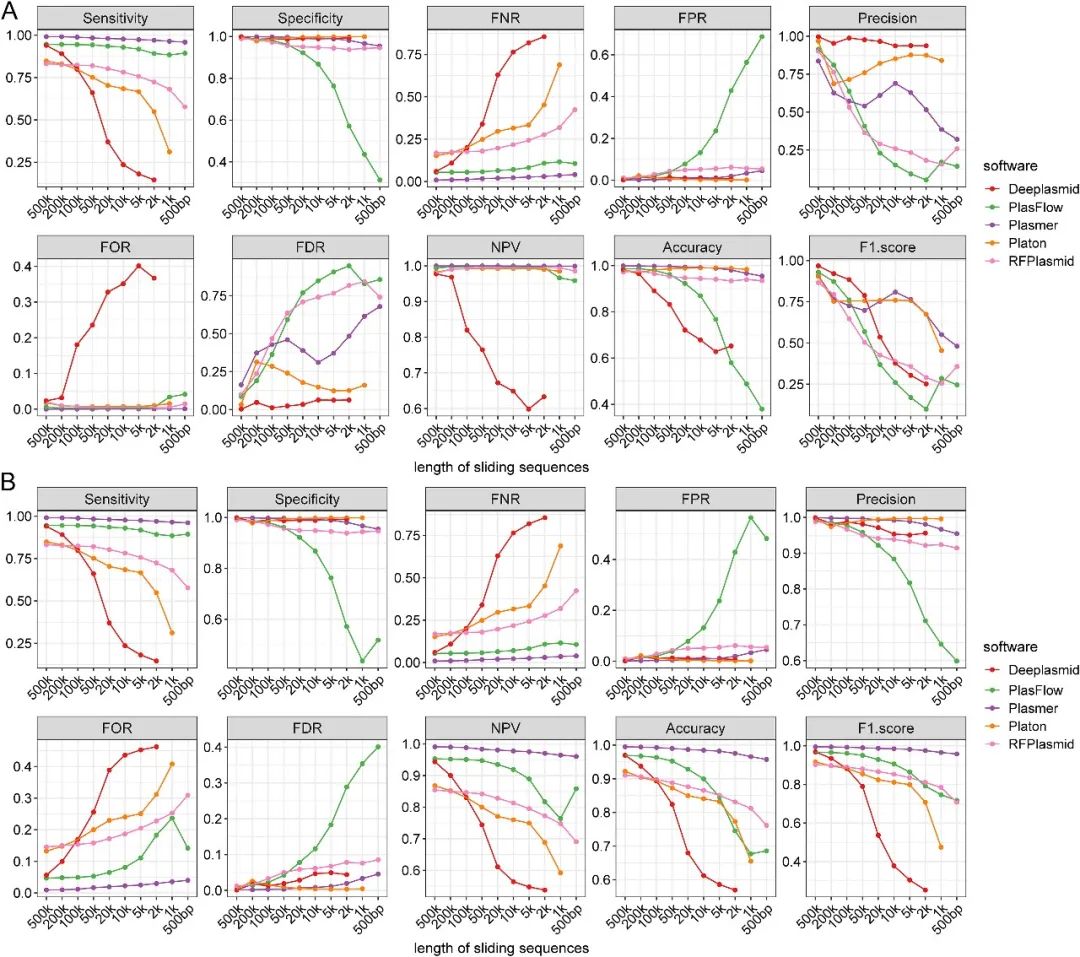
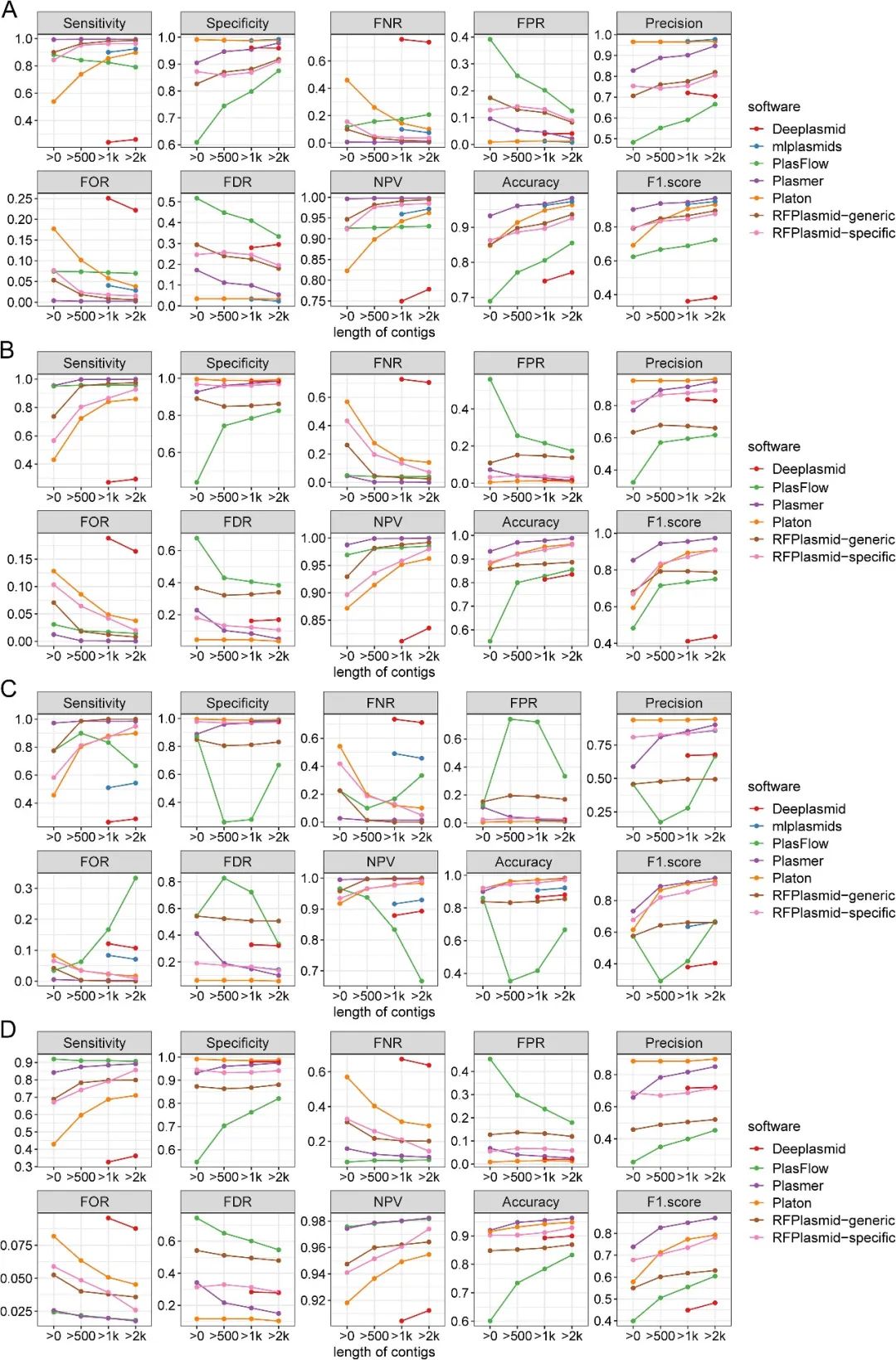
图2 基于随机森林分析的分类模型的性能 3. 使用剩余滑窗序列比较Plasmer和其他方法对不同输入长度输入序列的预测性能 Performance comparisons of different input lengths using remaining sliding sequences of all complete bacterial genomes in NCBI 为了评估 Plasmer 在不同输入长度上的性能和适用性,本研究使用除模型训练中使用的1,573,876条序列之外的其余不同长度的滑窗切分序列进行预测(500 kb、200 kb、100 kb、50 kb、20 kb、10 kb、5 kb、2 kb、1 kb和500 bp),计算每个长度的输入序列下,Plasmer与现有其他软件的性能参数,如敏感度、特异性、准确度、假阳性率及F1-score等等,综合所有参数比较现有软件与Plasmer的预测性能。 比较结果显示,对于所有测试长度,Plasmer均具有最高的灵敏度、NPV,以及与Platon类似的特异性、准确性和 FPR,以及最低的 FNR 和 FOR(见图3A)。值得注意的是,与其余软件相比,Plasmer 在灵敏度、特异性和准确性方面的表现始终更加稳定,即使输入序列长度降至500 bp,上述参数仍保持在0.95以上。 相比之下,Platon虽然具有和Plasmer相当的准确性、特异性和最低的FPR,但随着序列长度的降低,灵敏度和FNR明显下降。与Platon类似,RFPlasmid 具有良好的特异性和准确性,但灵敏度较差。PlasFlow在较长的序列上具有可接受的性能,但当序列短于10 kb时,准确性和特异性降低到 0.9 以下。相比于鉴定质粒序列,Deeplasmid似乎在鉴定染色体方面具有更好的性能。由于mlplasmids只有大肠杆菌、肺炎克雷伯菌和屎肠球菌三种预测模型,因此其不适用于滑窗切分序列的预测,不包含在此次比较当中。 整体来看,当输入序列长度降低时,大多数软件的灵敏度明显下降,这意味着预测结果中的“质粒缺失”(假阴性)。此外,一些方法对输入长度有限制。例如,Deeplasmid无法预测长度小于等于1 kb的序列,Platon也直接过滤掉所有短于1 kb的输入序列。因此,当输入序列长度下降到500 bp时,只有 Plasmer、RFPlasmid和PlasFlow 可以进行预测。 由于滑窗切分序列的数量随着长度的降低而急剧增加,导致染色体和质粒序列数量的极端不平衡,染色体的切分序列数比质粒切分序列数高出将近于10倍,这种不平衡会导致精确度、准确性、FDR 和 F1-score的偏倚。因此,通过将染色体序列下采样到与质粒序列数量一致,本研究进行了更有说服力的样本量平衡的测试。 在样本量平衡的测试中,Plasmer在大多数指标上均优于其他软件,具有最高的灵敏度、准确性、NPV和 F1-score,以及最低的 FNR 和 FOR(见图3B)。尽管Platon和 Deeplasmid在特异性、精确度、FPR 和 FDR 方面具有不错的性能,但随着输入长度的降低,它们的灵敏度和准确性迅速下降。即使在短序列上,Plasmer在所有指标上都表现出更加稳定的性能,而其他方法的性能则大幅度降低。 总之,对不同长度的滑窗序列的测试表明,Plasmer在染色体和质粒输入序列上均具有稳定的性能,并且是第一个可用于500 bp短片段的预测工具,这使得Plasmer成为稳定、准确和灵敏的质粒预测工具。
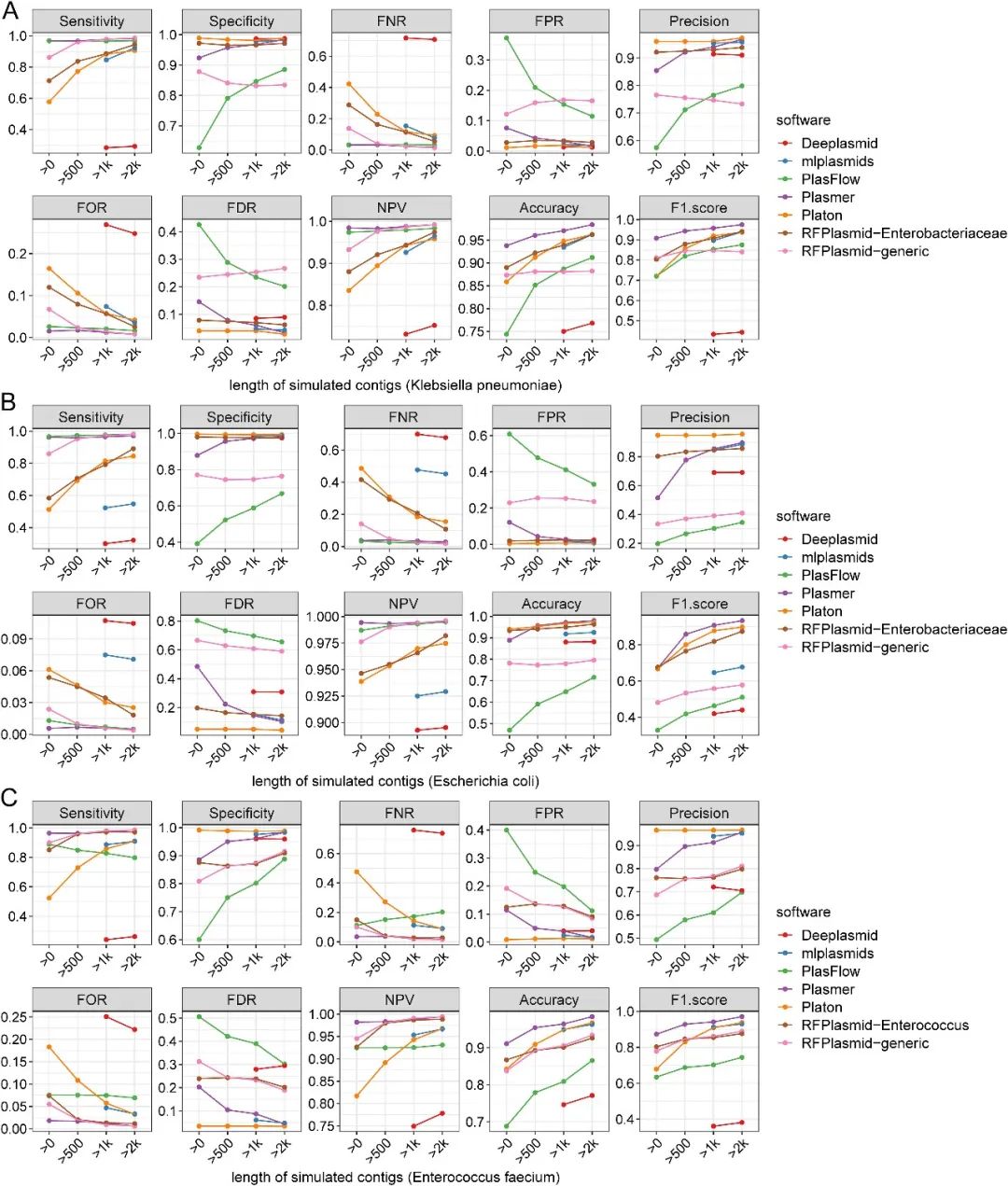
图3 Plasmer和其他方法在来自于NCBI中31,897个细菌完整基因组的剩余不同长度滑动序列的性能表现 4. 肺炎克雷伯菌、大肠杆菌和屎肠球菌模拟测序reads组装的性能比较 Performance comparison of simulated reads of K. pneumoniae, E. coli, and E. faecium 为了评估Plasmer在组装contigs上的性能,本研究分别模拟并重新组装了552 个肺炎克雷伯菌完整基因组、1718个大肠杆菌完整基因组和123个粪肠球菌完整基因组的测序数据。对于肺炎克雷伯菌,本研究标记了22,746个质粒和49,433 个染色体contigs。对于大肠杆菌,本研究标记了49,036个质粒和367,039个染色体contigs。对于屎肠球菌,本研究标记了9,490个质粒和20,357个染色体contigs。为了评估长度影响,本研究根据不同长度阈值对短contigs进行过滤(未过滤、500 bp、1 kb 和 2 kb),然后分别对过滤短contigs之后的序列进行预测。根据表3-9,本研究发现肺炎克雷伯菌和屎肠球菌的prevalence约为 0.3,大肠杆菌的prevalence仅为 ~0.15,这导致 Precision 和 FDR 失效,准确率和 F1-score 也会受到影响。相比之下,对于三个物种来说,Plasmer具有最高的准确性和 F1-score以及无偏的灵敏度和特异性(见图4)。虽然Platon的特异性和精确度比Plasmer稍高,但灵敏度下降到0.6以下,过滤到500 bp后仍低于0.8。对于RFPlasmid,本研究分别使用了通用和物种特异模型来进行预测,结果显示,物种特异模型的F1-score比通用模型更高,但是仍低于Plasmer。Mlplasmids性能均不理想。Deeplasmid 的特异性较高,但是敏感度极低。综上所述,对于模拟测序数据组装的contigs,Plasmer表现出更加均衡的性能,同时具有较高的敏感度和特异性。当去除短于500 bp的contigs时,Plasmer的灵敏度、特异性和准确性均高于 0.95,FPR 和 FDR 均低于 0.1。 5. 真实测序数据的性能比较 Performance comparisons using real sequencing data 为了评估 Plasmer 在真实测序数据上的性能,本研究收集了 mlplasmids文章中使用的41个屎肠球菌菌株和PlasFlow文章中使用的不同物种的40个菌株的原始测序数据。使用NCBI GenBank 中这些菌株的参考基因组进行验证。因为prevalence低于0.3,这意味着在真正的短读长组装结果中染色体序列数量比质粒更多,导致精确度、FDR、准确性和 F1 score产生偏差。结果表明,Plasmer具有最高的灵敏度、准确性、F1 score和NPV以及最低的FNR和FOR(见图5A,见图5B),这与模拟测序数据的评估结果相似。Platon具有较高的特异性,但不理想的敏感性导致其整体性能下降。相反,RFPlasmid具有出色的灵敏度,但特异性并不理想。Deeplasmid、mlplasmids 和 PlasFlow 等其他方法似乎在此类测试中表现不佳。以上测试表明,Plasmer 能够从短读长基因组组装中精确鉴定质粒或染色体片段,与其他现有方法相比具有更好的整体性能。
图4 Plasmer和其他方法在肺炎克雷伯菌、大肠杆菌和屎肠球菌模拟测序数据组装contigs的预测性能 为了评估 Plasmer 在没有参考基因组的新分离株上的性能,本研究下载了 Platon文章中使用的21个分离株的原始测序数据,每个分离株都有Nanopore长读长测序数据和Illumina短读长测序数据。本研究从头组装了Nanopore长读长测序数据,并标记了染色体和质粒序列,然后将它们作为“参考基因组”以验证各个工具对其相应的Illumina测序数据组装结果的预测。同时,由于这些菌株仍然属于大肠杆菌,为了剔除训练数据中大肠杆菌序列可能导致的偏差,本研究另外测试了Deeplasmid文章中使用的鲁氏耶尔森菌(Yersinia ruckeri)分离株,该物种在训练数据中不存在。由于没有可用于该耶尔森氏菌分离株的短读长测序数据,本研究根据Nanopore组装结果产生了模拟的短读长测序数据,并进行了测试。 如结果所示(见图5C),在对Platon文章中用到的21个菌株数据的测试中, Plasmer 仍然获得了最高的F1 score。在去除短于500 bp的序列后,Plasmer的特异性和准确性达到了0.95以上。Plasmer在未过滤的contigs上具有最高的 F1 score、灵敏度和NPV,最低的FNR和FOR。Platon具有最高的特异性和与Plasmer相当的准确性,F1 score仅次于Plasmer。然而,Platon的敏感性较差,这表明许多质粒序列被鉴定为染色体。RFPlasmid的“generic”模型具有与 Plasmer 相当的灵敏度、NPV、FNR 和 FOR,但与 Plasmer 和 Platon 相比,特异性、准确性和 F1 score较低且FPR较高。RFPlasmid的物种特异模型比“generic”模型表现更好,但其敏感度和F1 score仍低于Plasmer。尽管对于长于1 kb的序列来说, Deeplasmid特异性与 Plasmer 相当,但其灵敏度和F1 score较低。PlasFlow和mlplasmids在超过 1 kb 的contigs整体性能均不理想。
图5 Plasmer和其他方法在真实测序数据组装contigs上的预测性能 另外,对Yersinia ruckeri分离株的测试表明,即使prevalence低于0.1,Plasmer 也可以以最高的准确度和F1 score识别罕见物种中的质粒序列,这也证明了它在新物种中预测质粒序列的潜力。 上述结果证明了,无论对于已知物种或是未知物种,对于模型训练中不包含的基因组,Plasmer仍能够在从头组装的contigs中预测质粒序列,且准确性优于现有软件,具有广泛的适用性和出色的整体性能。 - 结论 - 综上所述,Plasmer是一种新型的细菌质粒预测方法,它具有超强的准确性和平衡的敏感性和特异性,即使在500bp以上的短序列上也具有最高的F1 score和稳定的性能。我们相信Plasmer的优势将使其成为比现有方法更可靠的选择,并促进相关研究,如水平基因转移和微生物抗性。 - 软件获取 - Plasmer是正在开发中的开源软件,可在https://github.com/nekokoe/Plasmer。在Docker Hub上有一个docker容器版本,网址是https://hub.docker.com/repository/docker/nekokoe/plasmer。Conda软件包也可在https://anaconda.org/iskoldt/plasmer;用户可以用 "conda install -c iskoldt -c bioconda -c conda-forge -c defaults plasmer "命令安装Plasmer。数据库可以按照GitHub的说明下载,也可以从Zenodo网站https://doi.org/10.5281/zenodo.7030674直接下载。 参考文献 Qianhui Zhu, Shenghan Gao, Binghan Xiao, Zilong He*, Songnian Hu*.Plasmer: an accurate and sensitive bacterial plasmid prediction tool based on machine-learning of shared k-mers and genomic features. Microbiology Spectrum. PMID: 37191574 - 通讯作者简介 -
中国科学院微生物所 胡松年 研究员,博士生导师 主要从事微生物宏基因组研究技术与方法、微生物多组学分析平台搭建以及微生物资源生物信息挖掘与利用,目前已在国际顶级期刊Science,Nature Plant, Genome Biology, Nature communications, Nucleic Acids Research等高水平SCI论文300余篇,主持或参与国家级研究项目二十余项。
北京航空航天大学 贺子龙 副研究员,PI,博士生导师 北航医工百人计划、热心肠智库专家、iMETA青年编委、中国健康管理协会植物营养与健康分会会员。致力于运用多组学手段揭示人体微生物与疾病之间的相互作用以及人体传染性疾病与致病微生物之间的联系。主持及参与中国博士后面上基金项目、北航医工百人计划、北航医工卓越创新计划、北航青年科技创新团队等项目,在Nucleic Acids Research, Briefings in Bioinformatics,Microbiology Spectrum,等期刊发表SCI论文二十余篇,总引用次数超1300次。 猜你喜欢iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAPiMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKlaiMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测 10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature 系列教程:微生物组入门 Biostar 微生物组 宏基因组 专业技能:学术图表 高分文章 生信宝典 不可或缺的人 一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote 扩增子分析:图表解读 分析流程 统计绘图 16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun 生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘 写在后面为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。 点击阅读原文 |



【本文地址】
今日新闻 |
推荐新闻 |