CMC专栏 |
您所在的位置:网站首页 › 空间设计目标 › CMC专栏 |
CMC专栏
|
来源:雪球App,作者: 博腾股份IR,(https://xueqiu.com/9430489416/189777792)
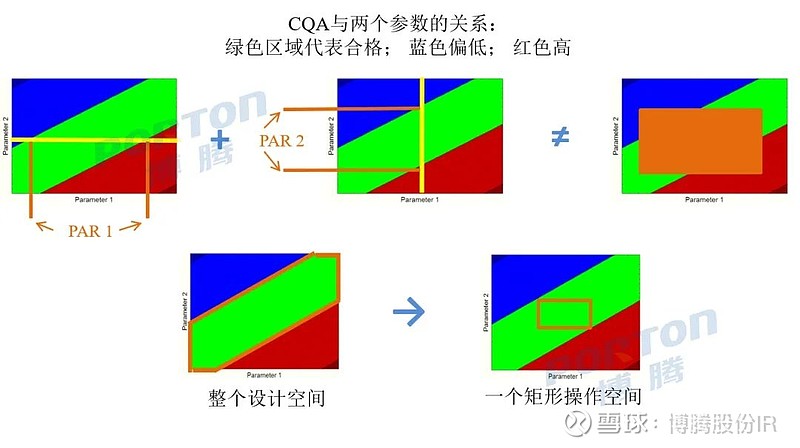
博腾微信公众号将不定期连载CMC(化学/生产/控制)相关文章,欢迎关注。本期文章约3000字。 设计空间 制剂研发QbD的终极目标(上) 一、概述 目前人们对QbD的认识日益加深。实施QbD理念的目的并不是消除商业化生产中人(人员操作)、机(设备)、料(原辅包)、法(生产工艺、工艺参数)、环(环境温湿度、微生物控制等)、测(分析方法)所导致的药品批内和批间的变异性,而是通过处方开发、工艺开发/验证和分析方法开发/验证分别开发出稳健的处方、生产工艺、分析方法以及相应的设计空间(Design Space),在GMP生产车间转化为能够落地的控制策略,从而确保能够持续地生产出质量稳定、具有足够工艺能力的药品。 设计空间的主题分为上下两篇,上篇重点介绍设计空间的分类、为什么要建立设计空间以及设计空间/控制空间与PAR / NOR的区别,下篇将重点介绍如何开发工艺设计空间、如何通过设计空间判断工艺稳健性。 二、设计空间的定义 ICH Q8对设计空间定义为“已被证明能保证产品质量的输入变量(如物料属性)和工艺参数的多维组合和交互作用的范围”。产品设计空间可分为处方设计空间、工艺设计空间和分析方法设计空间,本文主要讨论工艺设计空间。工艺设计空间是指一个可以生产出符合质量要求的参数空间。设计空间的优势在于为工艺控制策略提供一个更宽的操作面,在这个操作面内,物料的既有特性和对应工艺参数可以无需重新申请进行变化。 设计空间的研究是通过识别变异来源,通过生产过程设计管理变异性以及使用设计空间预测产品质量属性,促进对生产过程的理解。具体来说,工艺设计空间包括所有的单元操作、描述每个单元操作的过程参数以及所使用的原辅料,设计空间受所有工艺参数的可接受范围限制,包括关键工艺参数(CPPs)和非CPPs。由于设计空间的边界存在一定的不确定性,为保证能够持续可靠地生产出合格产品,往往在设计空间的范围内建立一个控制空间(控制空间为确保药品具有足够的工艺能力而在设计空间内划出范围更小的空间),用于保证工艺性能和产品质量的可靠性。 三、设计空间分类 狭义的设计空间仅指工艺设计空间,广义的设计空间还包括处方设计空间和分析方法设计空间。 处方设计空间 一般通过处方DoE研究设计获得,在设计空间中找出1-2个最优处方,并通过相容性试验、影响因素试验、3-6个月加速稳定性研究以及包材筛选研究确定最终处方。普遍认为处方一旦确定后即固定不变,因此不会涉及处方设计空间的概念,但考虑到处方风险/稳健性评价(如片剂处方中粘合剂和崩解剂用量的微小变化对溶出的影响)以及工艺放大过程中处方的可能变动(如片剂和胶囊处方中润滑剂和助流剂用量,缓释微丸处方中的包衣增重,HME处方中聚合物的用量),处方设计空间仍有重大意义。详细内容请参考此前文章“如何进行处方风险评价与获得稳健性处方”(文末链接跳转阅读) 工艺设计空间 一般所说的设计空间,本文将会重点讨论; 分析方式设计空间 在分析方法开发、优化、转移、验证过程中,主要通过Robustness研究中的DoE以及Ruggedness研究中的测量系统分析(MSA)开发出分析设计空间或方法可操作设计区域(MODR)。对于分析QbD的相关内容将在之后的文章中详细介绍。 按照制剂开发的不同阶段,可将工艺设计空间分为小试设计空间、中试/工艺放大设计空间、注册批次生产、预验证/验证设计空间以及生命周期管理设计空间,如下表所示。 开发阶段 目的 处方和小试工艺开发 通过小试工艺优化的DoE研究,开发小试批量条件下的设计空间、控制空间,通过QbD技术风险评价(TRA)确定该阶段的控制策略,并作为进一步中试放大/工艺放大的基础和先验知识。 中试工艺开发、工艺放大和优化 开发中试和注册批量条件下的设计空间、控制空间,通过技术风险评价(第1次Operational TRA)确定用于注册批次生产的控制策略。 注册批次(如NDA)生产 确认开发的设计空间和控制策略合理性,能够生产出具有一定工艺能力的药品。 预验证批次(如pre-PPQ)的生产 进一步开发和优化设计空间、控制空间,通过技术风险评价(第2次Operational TRA)确定用于验证批次和商业化生产的控制策略。 验证批次的生产(如PPQ) 确认开发的设计空间和控制策略合理性,能够生产出具有一定工艺能力的药品。 生命周期管理 定期开展技术风险评价,持续更新和优化设计空间和控制策略,持续改善药品工艺能力和质量。 四、建立设计空间的原因 一般选择建立设计空间的原因可分为以下3个方面 #1 ■单变量实验可用于范围研究以了解变量是否可能影响关键质量属性(CQAs),但一次仅涉及一个变量的实验不足以探索相互作用,输入变量和工艺参数可能对CQAs产生交互作用,应设计实验对相互作用进行研究并量化其影响,而设计空间便是知识空间中符合要求的输入变量与工艺参数的多维组合和交互作用的范围。在小试、中试、工艺放大、注册验证批次的不同阶段通过系统考察关键变量对iCQAs和CQAs的影响开发出相应的设计空间和控制策略,而设计空间考虑了多个工艺变量同时发生变动时对iCQAs和CQAs的影响,在上市后药品生命周期管理中通过持续积累批内和批间数据可持续更新和优化设计空间和控制策略,理论上可将药品质量和工艺能力提高到很高的水平,因此建立设计空间是一种更为有效的积累数据、信息和知识,持续加深工艺理解的有效方法。 #2 ■通过设计空间的大小和形状可判断工艺稳健性,并可通过模型模拟方法如蒙特卡洛模拟(下篇中将会介绍)预测连续商业化生产过程中药品的长期工艺能力。 #3 ■ 根据ICH Q8,设计空间是通过对知识空间的风险分析与试验设计而取得的,只要所有的控制空间都在设计空间的范畴内,那么控制策略在各个控制空间之间移动时,如材料的属性和工艺参数发生变化时,就无需进行进一步的注册审批,减少上市后的变更申报。这样制药企业就可以减少申报费用,节约时间,提高工艺改进的速度,最终达到增加利润的目的,同时降低了药监部门的监管和审批成本。 在以往大生产中,对很多工艺参数的控制很难做到一成不变,在申报生产工艺时,报的参数是一个点。如果在实际生产中不能准确控制参数,就会产生报废、返工甚至弄虚作假的现象。设计空间允许企业在研究的基础上确定一个可以保证产品质量的操作空间。开发适宜的设计空间注册申报方面更容易为药监部门接受,而适当的控制空间/控制策略更容易为生产质量系统所接受。制药企业通过建立设计空间,可减少药品生产的质量风险,减少生产成本,缩短投资回报时间,也减少监管部门工作量,对于制药企业和监管部门是一个双赢的结果。总体来说,设计空间赋予了注册审批灵活而科学的管理方式,建立设计空间是保证产品质量的一个强大工具,因此可以视为制剂研发人员的终极目标。 五、设计空间与PAR / NOR的区别 PAR(被证明的可接受范围)为在保持其它参数不变的前提下,对单个变量的范围进行考察(一般采用OFAT/EOF进行研究,具体方法可参见此前文章TRA部分,文末链接跳转阅读)。操作应在此范围内进行,当保持其他参数恒定时,在此参数范围内操作将生成符合相关质量标准的产品。 如下图,绿色区域代表建立的整个设计空间,由于PAR未评估输入(物料属性、工艺参数等)之间的关系和相互作用,由两个参数获得的PAR范围组合(橙色区域,其部分范围位于不合格区域)并不与设计空间完全等同。
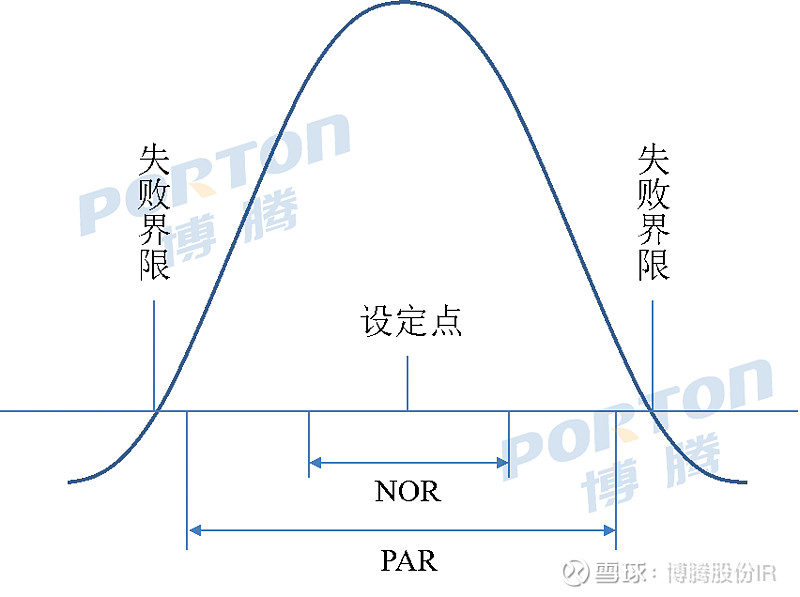
NOR(正常运行范围)描述的是目标操作条件邻近范围,其中包括正常的操作波动(波动并不总能控制)。 如下图,通过OFAT/EOF研究确定单个参数的PAR后,在其中间确定一个设定点,该设定点设置的一个邻近范围称之为NOR。NOR是在PAR范围内被定义的范围,用在生产操作指南中作为一个工艺参数能够被控制的目标和范围,在此范围内,生产单元操作的物料和最终产品能够符合放行标准和CQAs的要求。以往,我们只提交每个参数的NOR,在该范围以外的任何改变将表示批次失败,或者必须在变化实施之前提交并获批补充申请。而应用批准的设计空间,任何在NOR范围外而在设计空间范围内的变化均可接受。
未完待续。。。 下期预告 设计空间:制剂研发QbD的终极目标(下)
|





【本文地址】