Environmental Microbiology/ Venn图:寻找核心微生物组 |
您所在的位置:网站首页 › 物种分布分数 › Environmental Microbiology/ Venn图:寻找核心微生物组 |
Environmental Microbiology/ Venn图:寻找核心微生物组
|
背景 确定人类微生物群是否有核心是美国国立卫生研究院人类微生物群项目的目标。因此,许多研究旨在发现和比较与健康和疾病状态下的哺乳动物宿主相关的核心。这些关于核心的研究与主要类群同样引起人们极大的热情。核心的概念不限于与宿主相关的微生物群。环境微生物学家长期以来一直就自由生活的微生物群落提出类似的问题。短语“核心微生物群”可用于描述土壤、湖泊或废水处理系统中共享的成员。人类微生物项目重燃了经典问题的流行:这些核心微生物对群落有哪些作用? 2009年,Hamady 与Knight提出了关于人类个体核心性质的观点:核心可能是实质性的(其中大部分微生物类群是共享的);最小的、不存在的或梯度的(例如,沿着某种饮食或其他环境连续体)。他们还认为,一些核心只在宿主的亚种群中共享,而不是宿主物种的所有个体。这些深思熟虑和及时的想法为探索大型微生物组数据集提供了一个起点。尽管对核心的关注有所增加,但核心的构成仍然难以捉摸。一种典型的方法是基于存在/不存在数据集,研究从相似栖息地跨地点发现的物种数量。结果在维恩图的视觉上,圆圈代表不同的微生物群,它们的重叠区域代表核心。虽然维恩图是一个合理的初步探索,但它的概念基础忽略了生态特征,而生态特征可能暗示了一种更微妙的理解,即成员存在与他们在一种生态系统功能相关。 定义核心微生物组 核心通用定义导致数据集呈现为OTU表。OTU表有一行中的每个OTU(“物种”或感兴趣的单位)和群落观察(例如采样单位)。表格中充满了关于OTU事件的数字信息。OTU表不必局限于基于16S序列的宏基因组数据;它可以包括来自各种组学方法的数据,例如蛋白质或代谢物的丰度、与芯片上核酸探针的杂交强度或功能基因簇的存在/缺失。我们用微生物群来描述与栖息地相关的微生物群落,无论是活跃的还是不活跃的。栖息地是决定特定环境区域或地点的生物生态位空间的非生物组成部分。一个典型的观察是来自一个地方的一个时间点的一组微生物相关数据(例如,16S,转录组,宏基因组),但是相关的核心微生物群比较通常是在相似的栖息地进行的。微生物群落是微生物组的相互作用子集,是生态系统的活跃部分,包括生境中非生物和生物成分的相互作用动态。 成员:共享存在
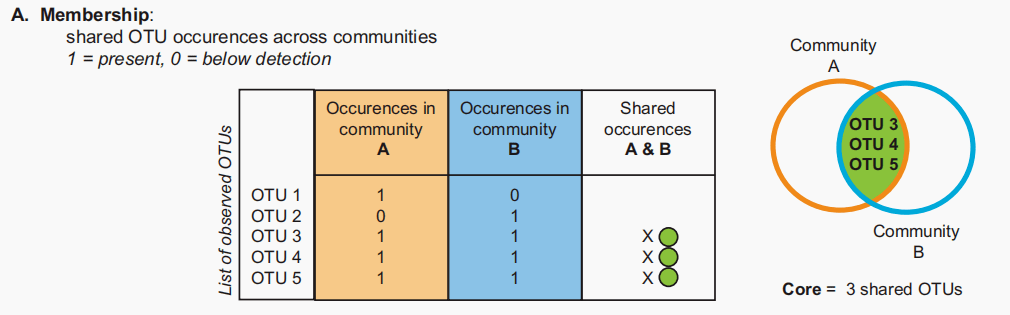
图A:基于共享成员的核心。输入OTU表是存在/不存在,共享存在的出现在感兴趣的群落中进行计数。 成员分析考虑了存在于两个或多个微生物群中的共享分类群(或基因)。这是基于存在/不存在数据集,其中共享的otu被计数(图1A)。维恩图的视觉显示了重叠的数量,但是对于多于四个类别组的表示无效。Sørenson指数说明了每组中共享的和唯一的otu的数量,也描述了基于成员的核心微生物群。 组成:优势、稀有和中间类群
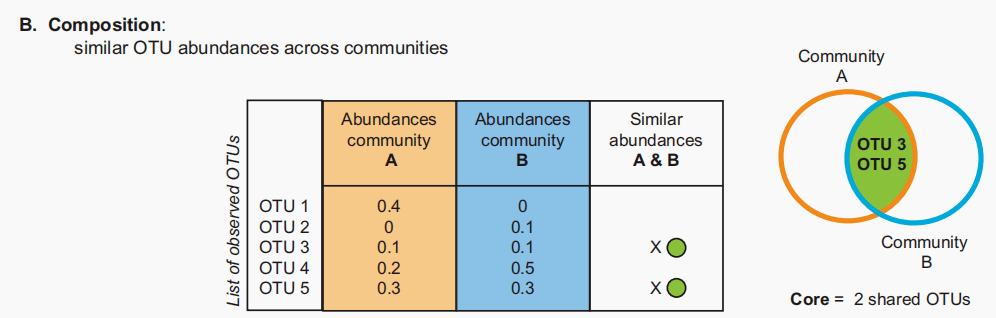
图B:基于共享组成的核心。在本例中,只有共享且比例相似的OTU才计入核心。 维恩图分析忽略了组成,这解释了每个OTU的相对丰度,探索微生物群落的多变量分析(多维标度或基于距离的分析)更加重视更丰富的otu,导致由最丰富的otu驱动的模式。相比之下,维恩分析对所有观察到的OTU进行同等加权,无论它们在群落中的代表性如何。 为了在核心分析中考虑组成,可以为每个微生物组创建秩丰度曲线(物种丰度分布)。可以进行有根据的“cut-offs”来分离最丰富和最不丰富的otu。传统生态学文献建议,最丰富的10%应该被认为是占优势的,最不丰富的65%应该被认为是稀有的。然而,微生物秩丰度曲线通常包括非常少的优势成员和许多稀有成员的长尾。这导致了一个高度“倾斜”的等级丰度分布,可能需要对优势、常见和罕见的otu进行不同的定义。物种多度分布的理论和应用在经典生态学文献中被积极讨论,这可以为优势和稀有的概念提供信息,并为微生物生态学中的定义提供指导。 系统发育和功能冗余
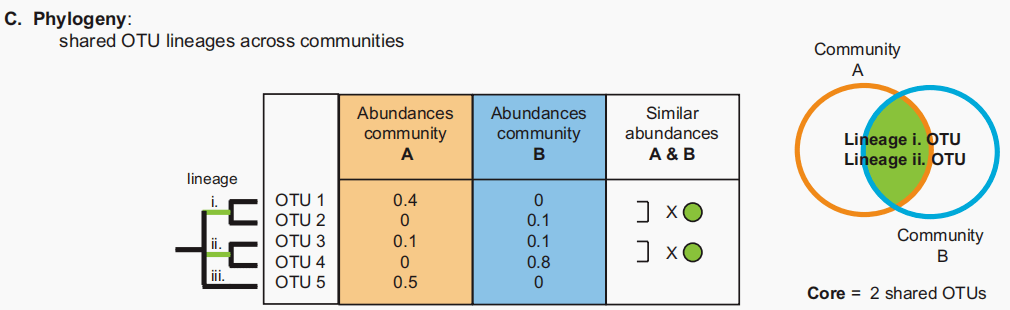
图C:包含系统发育信息的核心。相关的OTUs作为一个单元被计算到核心,就像谱系ii中的OTUs 1和2一样。系统发育水平可以上调或下调。 描述组成和功能的关系是微生物生态学的一个中心挑战。功能和组成在某些系统中是联系在一起的,但在另一些系统中却不是。此外,一些功能仅限于某些分类群(如硫酸盐还原),但其他功能在不同的群体中广泛存在(如光合作用)。微生物组可能包含系统发育和功能冗余。当来自同一谱系的多个OTU存在于一个微生物组中时,发生系统发育冗余,而当多个OTU在一个微生物组中执行相同的作用(例如固氮)时,发生功能冗余。谱系内部和之间的关系可以通过从组学数据集构建系统发生树来假设。当来自同一谱系的OTU也具有相似的功能时,系统发育和功能冗余可能重叠。然而,每种类型的问题都与计算多样性的建议相似:在核心的定义中,冗余的otu是否应该与唯一的otu同等计数(图1C)。 冗余对于定义和解释核心很重要。例如,干扰生态学假设一个强健的群落包含多个扮演相似角色的物种(如“保险假说”)。如果这个原则适用于微生物群落,多余的成员可以缓冲干扰反应。例如,如果不是所有功能冗余的成员都对抗生素敏感,那么在应用抗生素后,冗余可以使群落功能恢复。 微生物组的更多信息将为功能冗余的假说提供基础。在缺乏关于分类群功能作用的足够信息的情况下,系统发育信号可以预测OTUs的功能作用以及核心微生物组是否包含冗余。 持久性:微生物群动态
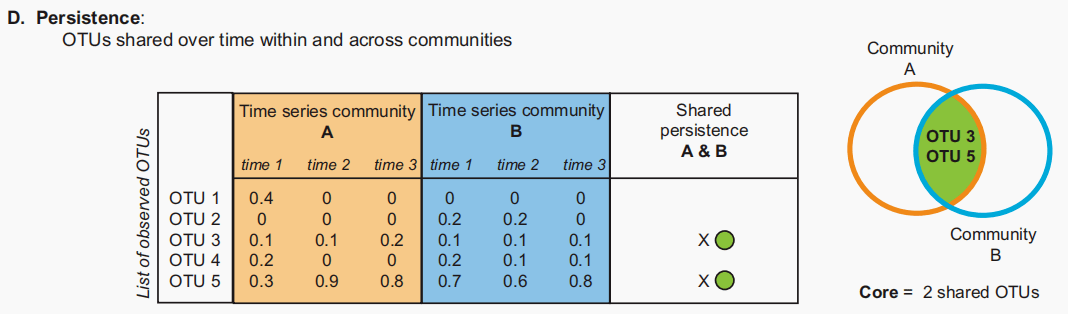
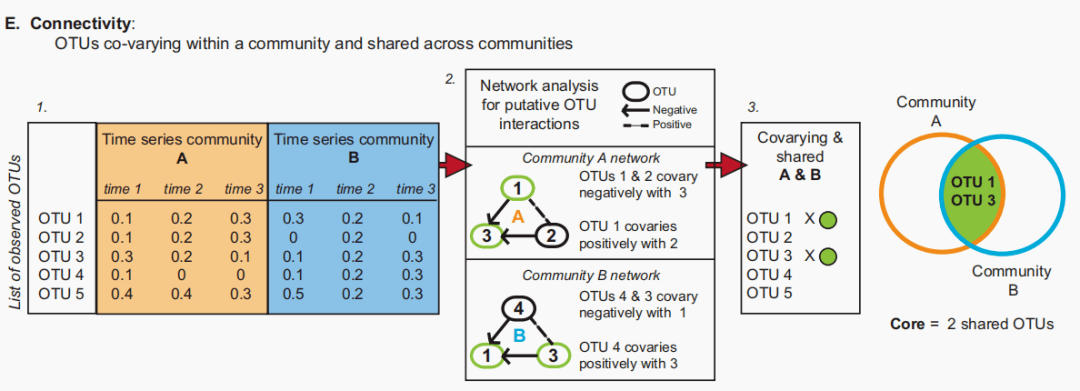
图D:核心微生物群,包括在一系列中持续存在的OTUs。在一个群落中持续观察到的otu被视为核心。(此处显示为每个群落A和B的连续时间点1、2和3)。 连通性:微生物互作
图E:一个核心,仅包括与其群落其他成员交互(或假定为交互)的OTU。重要的OTU相互作用的假设来自于对一系列的网络分析。在网络中,虚线代表假定的负相关(例如,物种竞争或捕食),带箭头的实线代表假定的正相互作用(例如,两个OTU都响应共享资源的增加)。 核心微生物群的概念也可以包括关于OTU假定的或证实的相互作用的信息。许多微生物学家和生态学家一致认为,一个群落是由一个区域内相互作用的物种组成的。这种鉴定是很重要的,因为任何基于DNA的序列分析都可以检测到无活性、死亡或暂时的OTUs。因此,通过连通性鉴定核心微生物群,如果没有生物相互作用的迹象,则省略分类群(图1E)。基于连通性的核心定义将包括在微生物组内具有假设的相互作用的微生物(揭示一个区域的微生物群落),但也在微生物组间共享(揭示相似栖息地的核心微生物组)。对此需要注意的是,一些关于缺乏连通性的预测是有缺陷的,因为可能存在应用于生态系统的方法未检测到的生物联系。 网络分析可以生成关于交互OTU的假设。网络在微生物生态学文献中越来越受欢迎,并且它们是微生物生态学工作流程中的标准,例如QIIME。网络分析有许多选项,包括一些从其他学科借用的选项。例如,将来自神经网络分析的自组织图应用于酸性矿山排水微生物群落,以了解强环境约束是否限制核苷酸组成。专门为微生物群落设计的网络分析较少,但一个例子是局部相似性分析(LSA),它是为了解海洋细菌群落的长期动态而创建的。LSA的两个优点是:(1)它使用了一个相关统计量,以适应otu之间的非线性关系,(2)可以纳入时间成分。这些对于揭示不一定是直接或瞬时的关系很重要,例如,争夺共享资源的两个种群之间的互补动态。针对微生物生态学问题进行严格的网络分析,并能够处理大型组学微生物数据集,这对于理解核心微生物群成员之间连通性的作用至关重要。在预测和管理整个系统的核心微生物群时,微生物群落内相互作用的数量或强度可能比otu的身份更具信息性。Gardner与 Ashby(1970)的经典工作表明,系统内的相互作用比例调节稳定性,但随着系统变得更大(更可能的相互作用),在超过“临界”相互作用比例后,它可能会突然变得不稳定。这将是一个有趣的概念,从昆虫肠道或酸性矿井排水群落到不同的哺乳动物肠道或热带土壤群落用不同复杂程度的微生物群进行测试。 注意事项和讨论要点 核心微生物群的所有这些定义都伴随着许多警告。实质性灵活性的一个来源是OTU定义。基于序列的OTU定义有多种比对和聚类算法,每种算法都有不同的偏差来源和方法误差容限。在许多情况下,共享otu的数量取决于序列同一性的程度或选择的分类水平。例如,如果在结构域水平定义,所有细菌群落将共享一个核心成员,相反,如果共享核心成员由99.9%的序列同一性定义,则没有两个群落将共享任何核心成员。在对14个环境栖息地的分析中,16S rRNA基因中98%、95%、92%和89%的核心序列同一性证据不足。在这项比较研究中,只有16%的otu出现在一个以上的栖息地的秩序水平。 定义OTUs的新方法可能会发现以前未检测到的核心微生物群。在最近的Meta分析中,揭示了肠道生境的核心微生物群,16S序列首先被翻译成五聚体频率以降低复杂性,然后这些频率通过主成分方法进行分析,并在排序空间中进行聚类以定义OTUs。通过将这种方法与16S rRNA基因系统发育分析进行比较,作者发现他们可以重建预期的多样性。他们的结果表明,使用多种方法来定义OTUs有助于探索核心微生物群。一个重大的挑战是决定哪种统计方法是最合适和最有生物学意义的。 揭露核心还有另外的注意事项。将每个OTU分类单元分配给一个分类单元会引入偏差,但随着每个测序计划的进行,数据库变得更加全面,这是由针对系统发育上代表性不足的微生物进行测序的项目(例如GEBA;http://www . jgi . doe . gov/programs/GEBA/index . html)。微生物研究技术会影响对核心的描述。以基于聚合酶链反应的测序为例,每次观察的reads、引物和测序区的选择以及测序深度将影响对核心的感知。最后,重复势在必行。如果在所有重复中没有一致检测到OTU,则不应将其包含在核心中。应该注意最好地理解每个偏差的来源,并相应地解释结果。定义核心的另一种方法是基于一组始终不存在而不是存在于微生物组或栖息地样本中的OTU。检测技术总是会限制对缺失数据的解释,因为缺失不能与存在但低于检测极限的OTU相区分。然而,随着技术的进步,群落的饱和度将会提高,对无遗漏地对不太复杂的微生物群进行采样的能力的信心可能会促进对共有缺失的评估。将功能性分析(功能性宏基因组学和基于芯片的分析)、过程分析(转录组学、蛋白质组学、代谢组学)和组学支持的分析(基因枪宏基因组学或16S rRNA基因测序)结合起来,将加深我们对共享的现有和缺失OTUs的重要性的理解。 结束语 最终,核心微生物群的适当定义取决于所解决的生态问题。因此,核心微生物群多重定义的应用将增强生态学的理解。维恩图只是开始。返回搜狐,查看更多 |
【本文地址】