单细胞测序最好的教程(七): 数据整合与批次效应校正 |
您所在的位置:网站首页 › 灯芯效应是在哪个流程产生的 › 单细胞测序最好的教程(七): 数据整合与批次效应校正 |
单细胞测序最好的教程(七): 数据整合与批次效应校正
|
作者按 本教程将是本系列教程中比较有趣的一章,对于大型的单细胞测序项目来说,数据整合也是不可或缺的一个步骤。本教程首发于[单细胞最好的中文教程](https://single-cell-tutorial.readthedocs.io/zh/latest/ ),未经授权许可,禁止转载。 全文字数|预计阅读时间: 5000|5min ——Starlitnightly 区别于我们以往所学的数据整合,在单细胞测序领域,数据整合涉及一个不得不去考虑的因素:样本的批次效应。 批次效应 批次效应是测量的表达水平的变化,这是处理不同组或“批次”中的细胞的结果。例如,如果两个实验室从同一队列中采集样本,但这些样本的解离方式不同,则可能会出现批次效应。如果实验室 A 优化其解离方案以解离样品中的细胞,同时最大限度地减少对细胞的压力,而实验室 B 没有这样做,那么 B 组数据中的细胞很可能会表达更多与压力相关的基因。 一般来说,批次效应的来源多种多样且难以确定。一些批次效应来源可能是技术性的,例如样品处理、实验方案或测序深度的差异,但供体变异、组织或采样位置等生物效应也可以被解释为批次效应。是否应考虑生物因素带来的批次效应取决于实验设计和所提出的问题。例如我们在探究肿瘤与性别的关系的时候,那么性别就应当保留,而不是当成批次效应去除。 1. 数据整合模型的类别在scRNA-seq中,消除批次效应往往由以下三个步骤构成: 降维建模并消除批次效应嵌入降维在上一章的描述中,我们提到了主成分分析法PCA,运用PCA可以降低我们数据的信噪比,提取出细胞的差异信息。而建模并消除批次效应则是核心步骤,绝大多数的批次效应校正算法都包括了如何构建一个稳定的数学模型。我们按照模型的发展顺序,可以分为四类:全局模型,线性嵌入模型,基于图的模型,深度学习模型 全局模型: 源自bulk RNA-seq,将批次效应建模为所有细胞中存在的(加法/或乘法)效应。一个常见的例子是 ComBat线性嵌入模型: 是第一个单细胞特异性批量去除方法。这些方法通常使用奇异值分解 (SVD) 的变体来嵌入数据,然后在嵌入中跨批次查找相似单元的局部邻域,并使用它们以局部自适应(非线性)方式校正批次效应。常见的例子包括最近邻MNN,Seurat,Harmony,Scanorama,FastMNN等基于图的模型: 通常是运行速度最快的方法。使用最近邻域图来表示每个批次的数据。通过强制连接不同批次的细胞,然后修建细胞类型组成的差异的图的边缘,可以纠正批次效应。一个常见的例子是BBKNN深度学习模型: 大多数深度学习批次效应校正方法都基于自动编码器网络,并且要么在条件变分自动编码器(CVAE)中对批量协变量进行降维,要么在嵌入空间中拟合局部线性校正。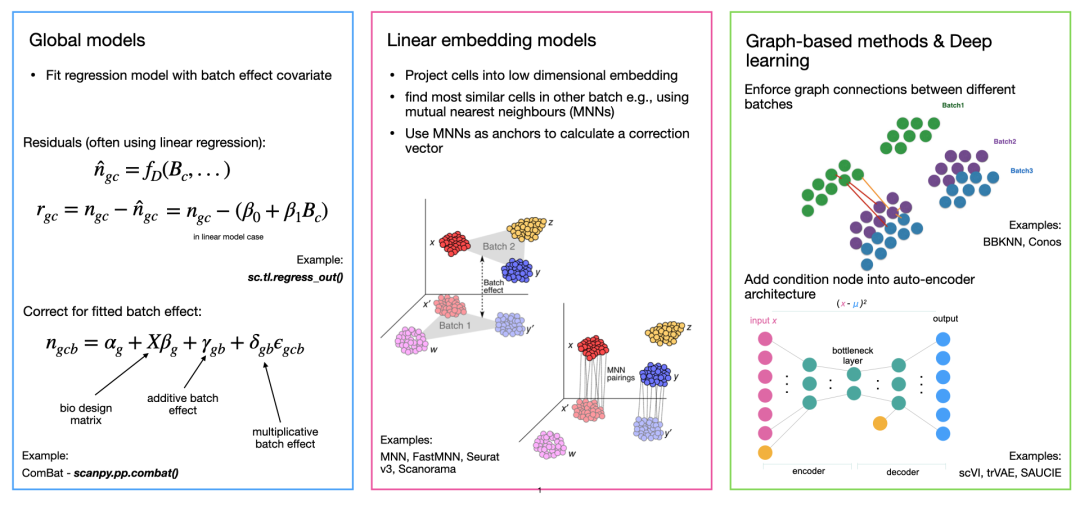 模型示例 2. 数据整合模型的比较在本教程中,我们将运行不同的批次效应算法来学习批次效应校正的过程,但是不同算法的比较在此前的研究中已经完成。一些基准测试评估了批次效应校正和数据集成方法的性能。当消除样本的批次效应时,方法可能会过度校正并消除除批次效应之外的有意义的生物变异。因此,除了消除批次效应外,方法是否保留了潜在的生物学意义,也是我们需要去着重考虑的一个点。 注意: 不存在适用于所有场景的最佳方法 对于一些简单的批次效应校正任务来说,Seurat及Harmony的表现始终不错,而对于一些复杂的批次效应校正任务来说,scVI、scGen、scANVI和Scanorama则表现地更为出色。我们可以使用scib包来评估数据整合的效果,scib包中包含了10个指标,我们会在下面的探讨中详细介绍,让我们先导入我们的分析环境。 代码语言:javascript复制import omicverse as ov print(f"omiverse version: {ov.__version__}") import scanpy as sc print(f"scanpy version: {sc.__version__}") import scvi print(f"scvi version: {scvi.__version__}") import scib print(f"scib version: {scib.__version__}") ov.ov_plot_set() 代码语言:javascript复制 # omiverse version: 1.4.17 # scanpy version: 1.9.1 # scvi version: 0.20.3 # scib version: 1.1.4 3. 导入数据我们分析所用到的数据中包括了来自三个不同样本的骨髓数据,该数据集最早发布于2021年的单细胞分析 NeurIPS 竞赛[1]。我们从中选择了我们选择了 s1d3、 s2d1和 s3d7的样本进行集成,数据可以从figshare下载 s1d3: https://figshare.com/ndownloader/files/41932005s2d1: https://figshare.com/ndownloader/files/41932011s3d7: https://figshare.com/ndownloader/files/41932008我们读取这三个数据,并进行合并。 代码语言:javascript复制#读取数据 adata1=ov.read('neurips2021_s1d3.h5ad') adata1.obs['batch']='s1d3' adata2=ov.read('neurips2021_s2d1.h5ad') adata2.obs['batch']='s2d1' adata3=ov.read('neurips2021_s3d7.h5ad') adata3.obs['batch']='s3d7' #合并数据集 adata=sc.concat([adata1,adata2,adata3],merge='same') adata.obs['batch'].unique() 代码语言:javascript复制 #array(['s1d3', 's2d1', 's3d7'], dtype=object) 代码语言:javascript复制adata=sc.read('neurips2021_scib.h5ad')对于该数据,我们可以发现其有很多个obs的值,其中cell_type和batch是我们所关注的。 cell_type: 每个barcode的细胞类型batch: 每个barcode所处的批次对于现实的分析,我们可能需要考虑更多的变量,但在本教程中,为了简单起见,我们在这里只考虑batch。 我们首先对数据进行预处理,这里包括质控,归一化,高可变基因的筛选,与前面四个章节不同的是,对于数据整合的质控,我们提供了参数batch_key来进一步减少数据误差以保证质控的可靠性。 代码语言:javascript复制#质控 adata=ov.pp.qc(adata, tresh={'mito_perc': 0.2, 'nUMIs': 500, 'detected_genes': 250}, batch_key='batch') #存放原始数据 ov.utils.store_layers(adata,layers='counts') #归一化/高可变基因筛选 adata=ov.pp.preprocess(adata,mode='shiftlog|pearson', n_HVGs=3000,batch_key='batch') adata我们只保留高可变基因,在后续的批次效应校正中,也只需要用到高可变基因,不过不同的方法需要的数据可能会有所区别,例如归一化值/原始值的区别。 代码语言:javascript复制adata.raw = adata adata = adata[:, adata.var.highly_variable_features] 4. 批次效应可视化通过前面的讲述,你可能已经对批次效应有了一些概念上的理解。但是这些理解还不够直观,还比较抽象。因此,我们使用嵌入来直观感受一下数据中存在的批次效应。 代码语言:javascript复制ov.pp.scale(adata) ov.pp.pca(adata,layer='scaled',n_pcs=50) adata.obsm["X_mde_pca"] = ov.utils.mde(adata.obsm["scaled|original|X_pca"])我们发现数据中存在着非常明显的批次效应,对于CD14+ Mono,细胞被分为了三个不同的区域,分别属于三个不同的样本来源,但实际上三个样本的队列是相同的。 代码语言:javascript复制ov.utils.embedding(adata, basis='X_mde_pca',frameon='small', color=['batch','cell_type'],show=False)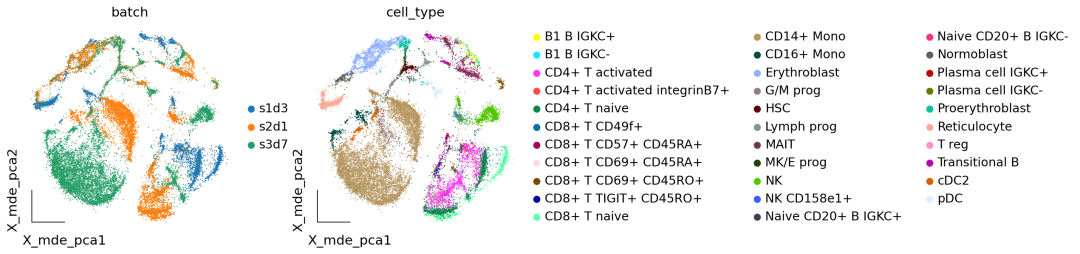 未去除批次效应的样本分布 5. 全局模型我们要尝试的第一个批次效应的算法是基于全局模型的combat,我们在omicverse中重载了该算法,并提供了简单的函数借口进行调用。关于combat的更多介绍感兴趣的读者可以去阅读相关的论文。 代码语言:javascript复制adata_combat=ov.single.batch_correction(adata,batch_key='batch', methods='combat',n_pcs=50) adata 代码语言:javascript复制 # Standardizing Data across genes. # Found 3 batches # Found 0 numerical variables: 代码语言:javascript复制 # Fitting L/S model and finding priors # Finding parametric adjustments # Adjusting data我们可视化Combat后的批次效应校正结果。我们发现样本中还是存在着比较明显的批次效应,这表明Combat可能不太适用于我们的骨髓数据集。 代码语言:javascript复制adata.obsm["X_mde_combat"] = ov.utils.mde(adata.obsm["X_combat"]) ov.utils.embedding(adata, basis='X_mde_combat',frameon='small', color=['batch','cell_type'],show=False)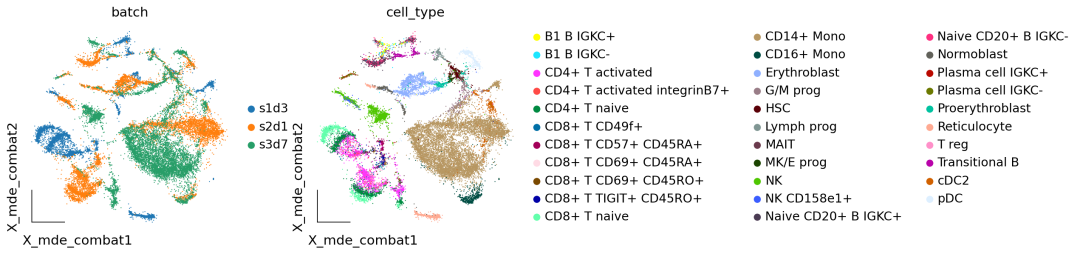 Combat去除批次效应效果 6. 线性嵌入模型线性嵌入模型我们一共会使用Harmony与scanorama来进行演示。我们首先介绍的是Harmony 6.1 HarmonyHarmony,是一种稳健、可扩展且灵活的多数据集集成算法,可满足无监督 scRNAseq 联合嵌入的四个关键挑战:扩展到大型数据集、识别广泛群体和细粒度子群体、适应复杂实验的灵活性设计以及跨模式整合的能力。 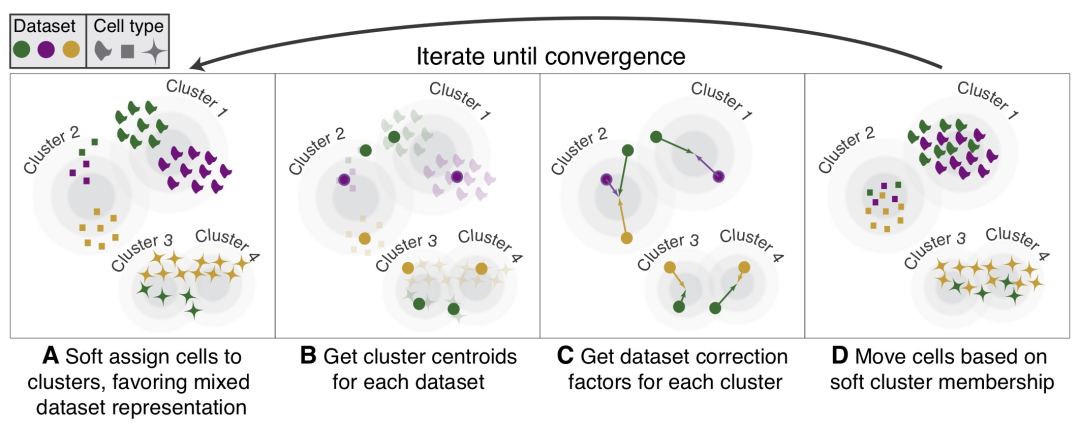 Harmony 代码语言:javascript复制adata_harmony=ov.single.batch_correction(adata,batch_key='batch', methods='harmony',n_pcs=50) 代码语言:javascript复制# ... as `zero_center=True`, sparse input is densified and may lead to large memory consumption # 2023-08-10 21:03:03,599 - harmonypy - INFO - Computing initial centroids with sklearn.KMeans... # 2023-08-10 21:03:09,288 - harmonypy - INFO - sklearn.KMeans initialization complete. # 2023-08-10 21:03:09,444 - harmonypy - INFO - Iteration 1 of 10 # 2023-08-10 21:03:15,470 - harmonypy - INFO - Iteration 2 of 10 # 2023-08-10 21:03:21,530 - harmonypy - INFO - Iteration 3 of 10 # 2023-08-10 21:03:28,488 - harmonypy - INFO - Iteration 4 of 10 # 2023-08-10 21:03:34,733 - harmonypy - INFO - Iteration 5 of 10 # 2023-08-10 21:03:41,025 - harmonypy - INFO - Iteration 6 of 10 # 2023-08-10 21:03:47,371 - harmonypy - INFO - Iteration 7 of 10 # 2023-08-10 21:03:53,738 - harmonypy - INFO - Iteration 8 of 10 # 2023-08-10 21:03:59,959 - harmonypy - INFO - Iteration 9 of 10 # 2023-08-10 21:04:06,248 - harmonypy - INFO - Iteration 10 of 10 # 2023-08-10 21:04:11,180 - harmonypy - INFO - Stopped before convergence我们继续使用嵌入可视化来观察Harmony的结果,我们发现CD4+ Mono细胞中,s3d7和s2d1被很好地混合到了一起,看起来,样本的批次效应得到了很好的校正。 代码语言:javascript复制adata.obsm["X_mde_harmony"] = ov.utils.mde(adata.obsm["X_harmony"]) ov.utils.embedding(adata, basis='X_mde_harmony',frameon='small', color=['batch','cell_type'],show=False)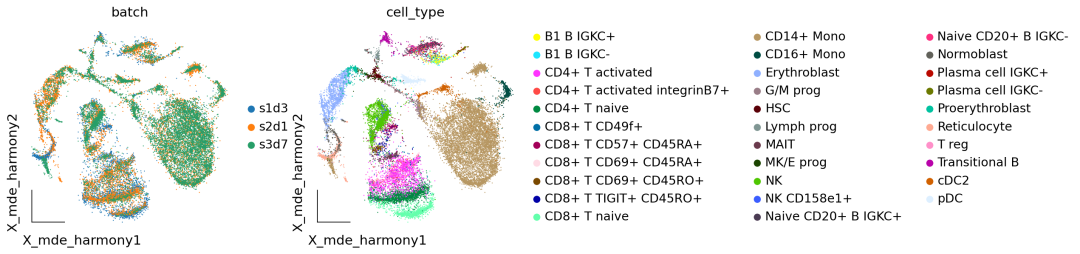 harmony去除批次效应效果 6.2 scanoramaScanorama可以识别和合并共享的细胞类型之间的所有数据集和准确集成的 scRNA-seq 数据的异构集合。相似地,我们在omicverse也实现了scanorame的使用,也是通过batch_correction函数。  scanorama 代码语言:javascript复制adata_scanorama=ov.single.batch_correction(adata,batch_key='batch', methods='scanorama',n_pcs=50) 代码语言:javascript复制 # s1d3 # s2d1 # s3d7 # Found 13953 genes among all datasets # [[0. 0.47831224 0.37535865] # [0. 0. 0.75730994] # [0. 0. 0. ]] # Processing datasets (1, 2) # Processing datasets (0, 1) # Processing datasets (0, 2) # (26970, 50)我们发现经过scanorama后的样本,批次效应也得到了很好地改善,那么Harmony和scanorama谁的效果会更好一些呢?我们在后面的章节中会对比不同的嵌入的效果,让我们继续看下一个 代码语言:javascript复制adata.obsm["X_mde_scanorama"] = ov.utils.mde(adata.obsm["X_scanorama"]) ov.utils.embedding(adata, basis='X_mde_scanorama',frameon='small', color=['batch','cell_type'],show=False)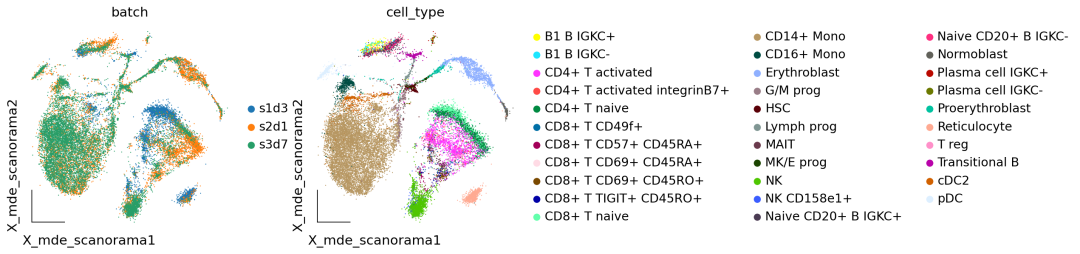 scanorama去除批次效应效果 7. 深度学习模型7.1 基于变分自编码器的模型scVI(单元变分推理),这是一种基于条件变分自动编码器的方法[ Lopez等人,2017]。,2018 ]可在scvi-tools包中使用[ Gayoso等人。,2022 ]。变分自动编码器是一种试图降低数据集维数的人工神经网络。条件部分是指在特定协变量(在本例中为batch)上调节此降维过程,以便协变量不会影响低维表示。在基准研究scVI已被证明在一系列数据集上表现良好,在批次校正之间取得了良好的平衡,同时保留了生物变异性[ Luecken等人。,2021 ]。scVI直接对原始计数进行建模,因此为它提供计数矩阵而不是归一化表达矩阵非常重要。 代码语言:javascript复制adata1=adata.copy() scvi.model.SCVI.setup_anndata(adata1, layer="counts", batch_key="batch")scVI创建的字段以_scvi. 这些是专为内部使用而设计的,不应手动修改。scvi-tools作者的一般建议是在模型训练完成之前不要对我们的对象进行任何更改。在其他数据集上,您可能会看到有关包含非标准化计数数据的输入表达矩阵的警告。这通常意味着您应该检查提供给设置函数的层adata.layers是否确实包含Counts。但如果您对来自全长协议或来自另一种不产生的定量方法的数据执行基因长度校正而获得的值,也可能会发生这种情况整数计数。 代码语言:javascript复制vae = scvi.model.SCVI(adata1, n_layers=2, n_latent=30, gene_likelihood="nb") vae 代码语言:javascript复制# SCVI Model with the following params: # n_hidden: 128, n_latent: 30, n_layers: 2, dropout_rate: 0.1, dispersion: gene, gene_likelihood: nb, # latent_distribution: normal # Training status: Not Trained # Model's adata is minified?: False我们在准备好数据与模型后,就开始训练了,默认情况下,scVI使用以下启发式方法来设置纪元数。对于细胞数少于 20,000 个的数据集,将使用 400 个 epoch,随着细胞数量增加到 20,000 以上,epoch 的数量会不断减少。这背后的原因是,当模型在每个epoah看到更多的barcodes时,它可以学到与从更多batch和更少的barcodes的信息中学到相同的信息量。 代码语言:javascript复制vae.train() 代码语言:javascript复制 # INFO: GPU available: True (cuda), used: True # INFO:lightning.pytorch.utilities.rank_zero:GPU available: True (cuda), used: True # INFO: TPU available: False, using: 0 TPU cores # INFO:lightning.pytorch.utilities.rank_zero:TPU available: False, using: 0 TPU cores # INFO: IPU available: False, using: 0 IPUs # INFO:lightning.pytorch.utilities.rank_zero:IPU available: False, using: 0 IPUs # INFO: HPU available: False, using: 0 HPUs # INFO:lightning.pytorch.utilities.rank_zero:HPU available: False, using: 0 HPUs # INFO: LOCAL_RANK: 0 - CUDA_VISIBLE_DEVICES: [0] # INFO:lightning.pytorch.accelerators.cuda:LOCAL_RANK: 0 - CUDA_VISIBLE_DEVICES: [0] # Epoch 297/297: 100%|█████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 297/297 [06:56 |
【本文地址】
今日新闻 |
推荐新闻 |