蛋白分析 |
您所在的位置:网站首页 › 氨基酸序列推断题方法有哪些 › 蛋白分析 |
蛋白分析
|
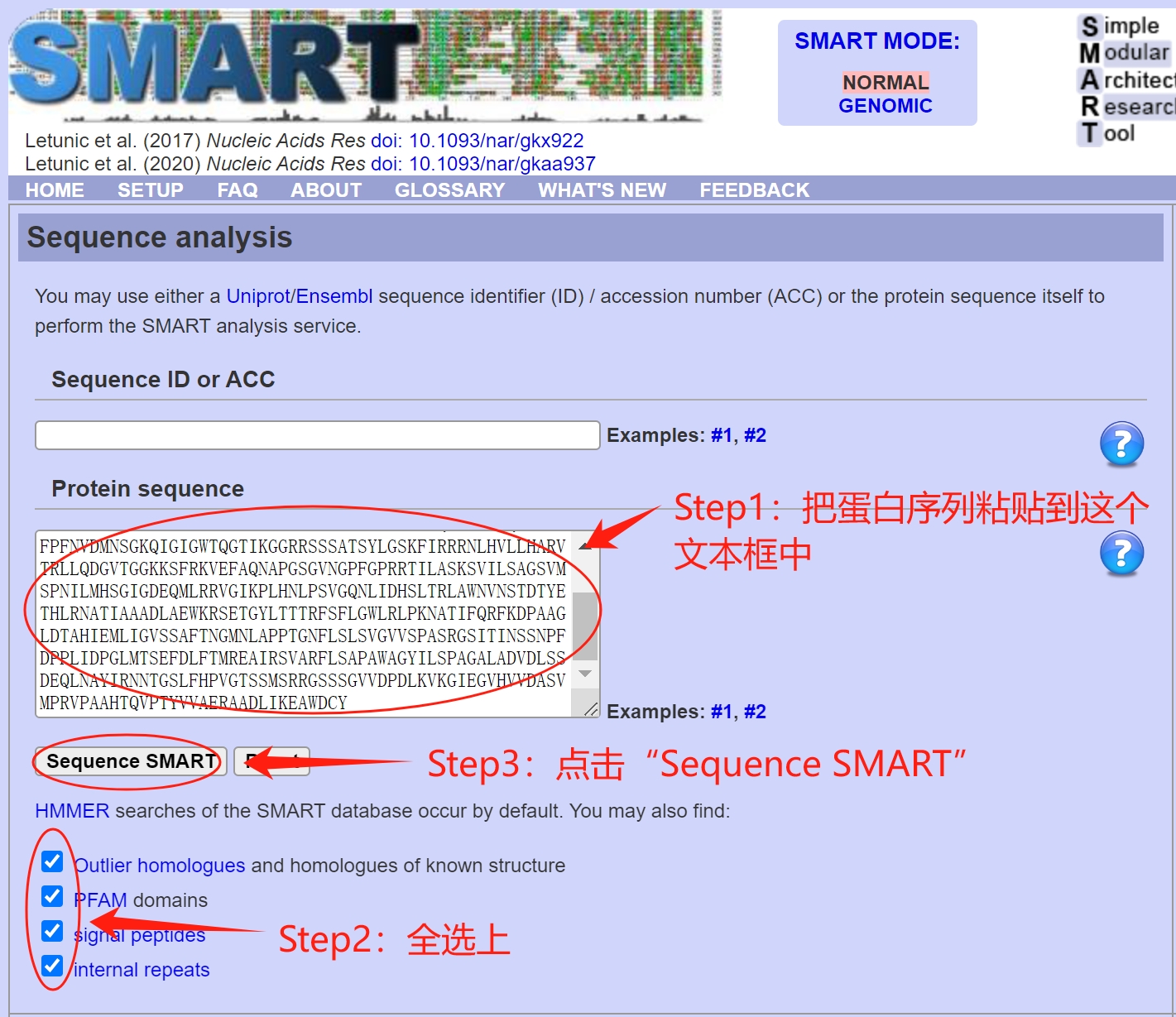
原创内容,欢迎转载,转载请注明出处** 主笔:于浩 注意事项 能够进行蛋白结构域预测的网站和工具有很多,最常用的数据库就是SMART数据库、NCBI的CDD数据库、InterPro数据库、Pfam数据库,这些数据库有自己的独特的地方,也有统一性,比如说前面几个数据库都整合了Pfam的结构域。具体的详细的信息可以自己查阅文献或者资料。 对于单个蛋白的分析建议使用SMART数据库,快速,图形化界面友好。NCBI的CDD数据库非常全,提供的信息很多。自己用Hmmer工具进行pfam数据库检索的好处是结果可以整理成为标准的格式,便于后续的分析。 利用SMART数据库进行蛋白结构域预测 单个蛋白的结构域分析1、进入SMART数据库进行检索 在文本框中输入蛋白序列(可以只有氨基酸序列,也可以以fasta格式粘贴),把下面的要鉴定的复选框都选上,点击“Sequence SMART”就可以进行蛋白结构域分析了,速度非常快。
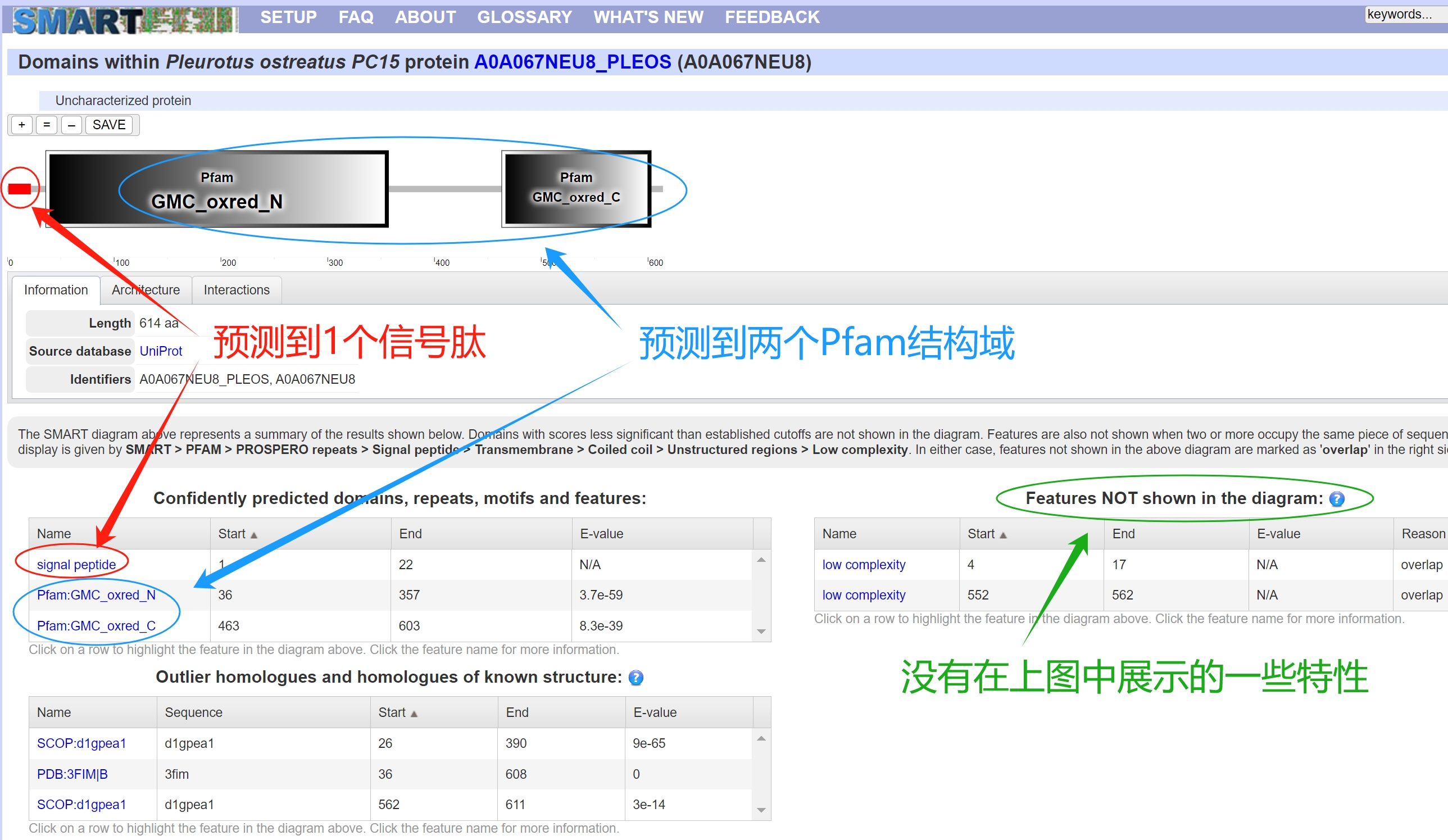
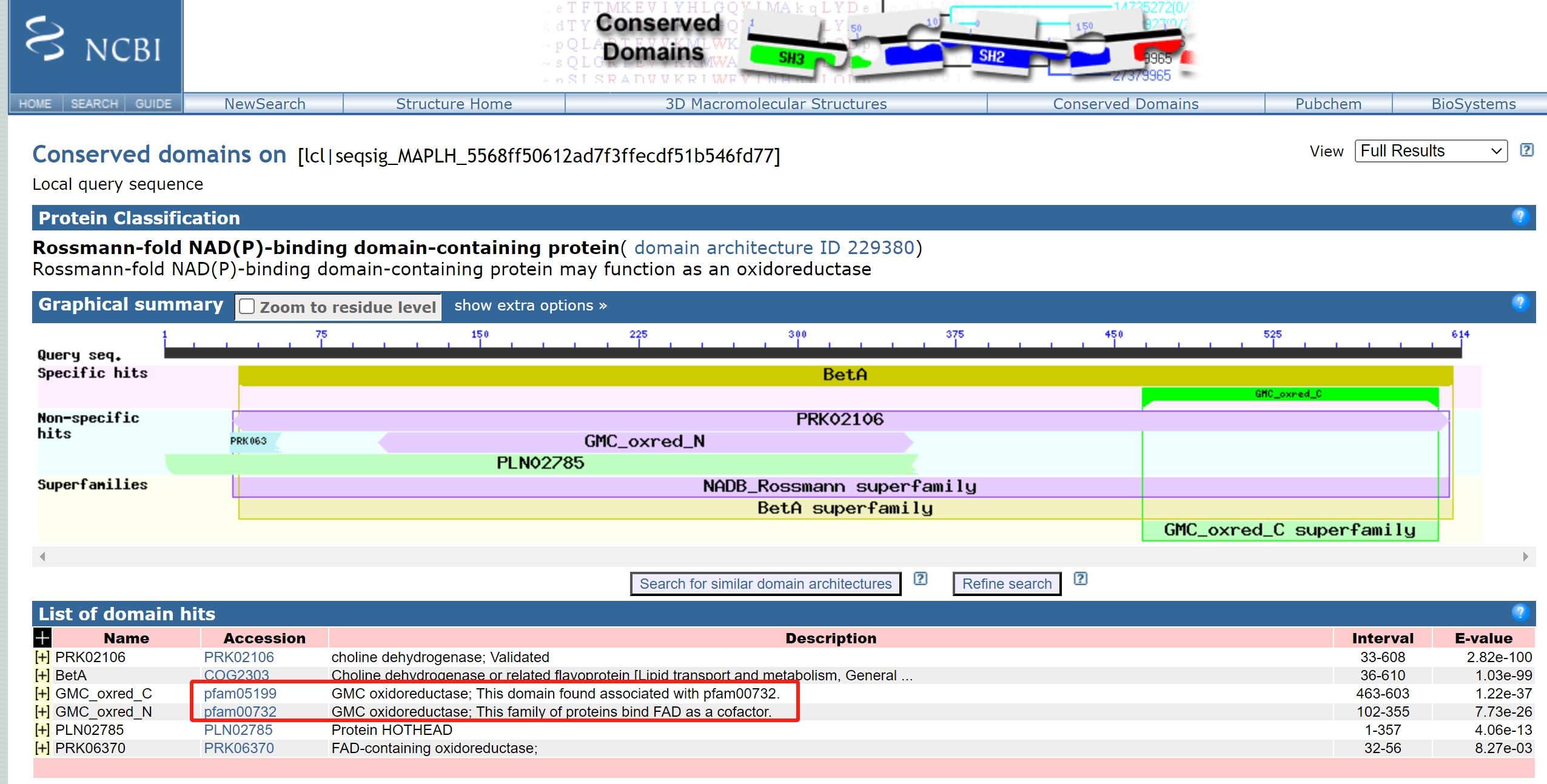
2、结果展示 分析完成后会弹出一个新的窗口,里面有预测的结果,可以看到下面的蛋白预测到了一个信号肽signal peptide,预测到了2个Pfam结构域分别是“GMC_oxred_N"结构域和”GMC_oxred_C“结构域。
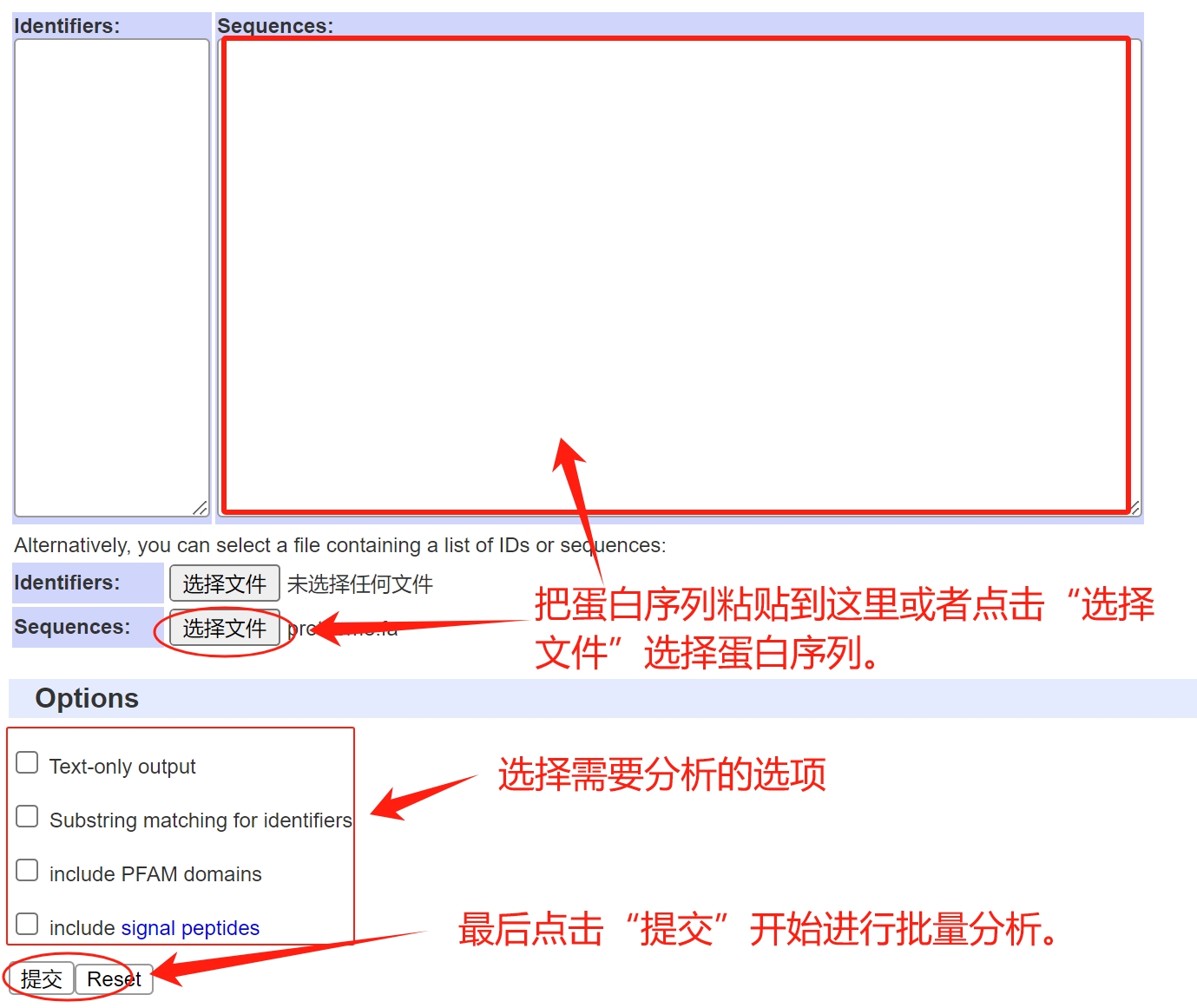
点击下面的网址链接,进入批量分析界面:http://smart.embl.de/smart/batch.pl 在网页的“Sequences”文本框中输入蛋白序列,或者点击Sequences”后面的“选择文件”选择fasta格式的蛋白序列文本文档。 选择完成后把Options前面的复选框都选上。 点击“提交”,开始分析。
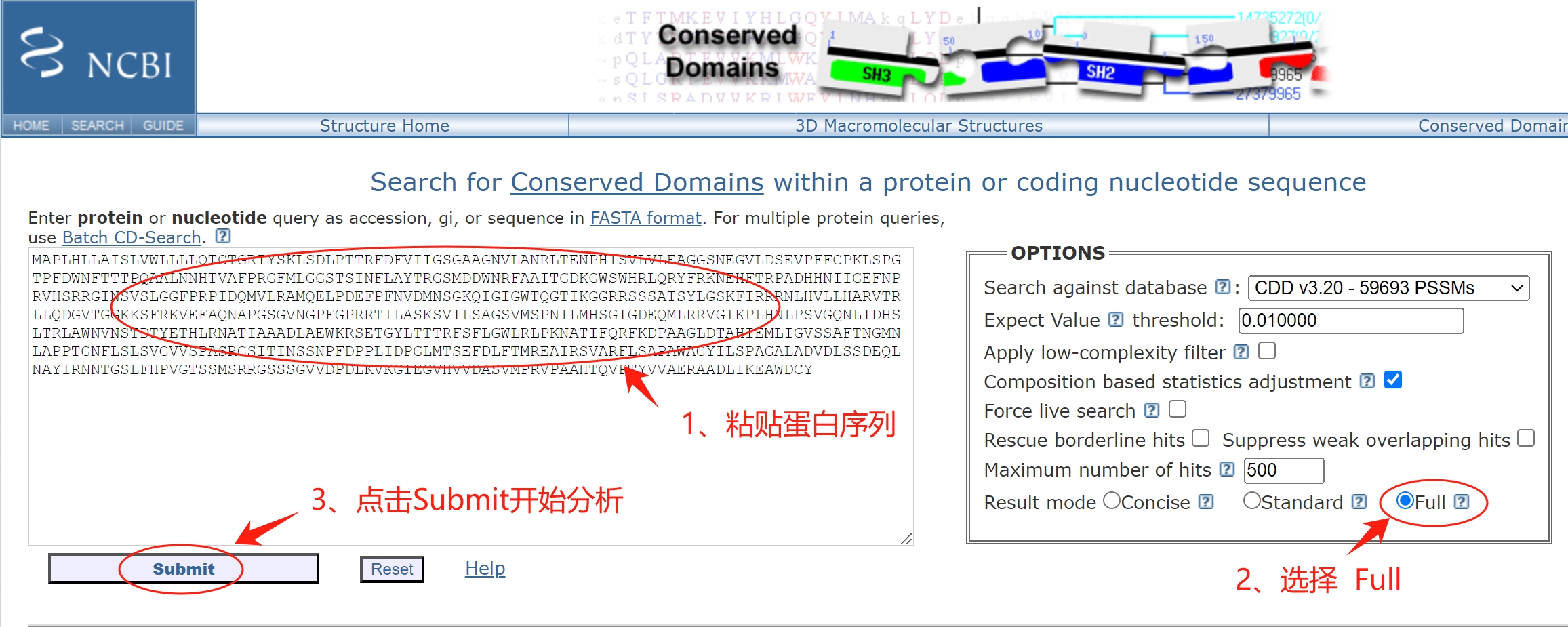
1、进入CDD数据库进行检索 点击网址链接进入比对界面:https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi 跟SMART一样,输入蛋白序列,选择要分析的内容,点击“submit”开始分析。
2、结果展示 下面的结果我们应该都比较熟悉,利用NCBI的Blastp比对蛋白的小伙伴都应该看过这个结果。 看起来要比SMART整合了更多的数据库的信息,NCBI牛!
批量比对的网址如下:https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi 这里就不展示教程了,是时候展示你自己强大的自学能力了! Hmmer软件利用Pfam数据库进行蛋白结构域预测 准备工作 1、下载Pfam数据库 Pfam数据库的下载地址如下:http://ftp.ebi.ac.uk/pub/databases/Pfam/releases/ 下拉窗口,可以看到最新的Pfam数据库(2024年1月份是36.0),点击进入文件夹,右键点击“Pfam-A.hmm.gz“,点击”复制链接地址“。 在Linux系统中利用wget工具下载Pfam数据库。 # 下载Pfam数据库 wget http://ftp.ebi.ac.uk/pub/databases/Pfam/releases/Pfam36.0/Pfam-A.hmm.gz # 解压缩数据库 gzip -d Pfam-A.hmm.gz2、在Linux系统中安装HMMER工具 建议使用系统工具或者Conda安装Hmmer,具体的代码如下 ubuntu系统中使用apt-get安装: apt-get install hmmer使用conda安装代码: conda install hmmer也可以自己下载安装这个软件,具体的方法如下。 # 首先下最新的hmmer软件压缩包(改地址可以下载到最新版本,也可以上官网自行下载最新版的压缩包) wget http://eddylab.org/software/hmmer/hmmer.tar.gz # 解压缩 tar -zxvf hmmer.tar.gz # 进入文件夹(我2024年1月份下载的是3.4版本) cd hmmer-3.4/ # 编译 ./configure # 编译 make # 最后一步添加环境变量 批量序列比对和信息提取1、Pfam蛋白结构域预测 准备好了Pfam数据库和hmmer工具之后,把所有的蛋白序列放到proteome.fa文本文档里面,可以用下面的代码来进行蛋白批量的pfam结构域预测。 # 用下面的脚本批量进行蛋白的pfam结构域预测 hmmscan -o out.txt --tblout pfam_out.tbl --noali -E 1e-5 Pfam-A.hmm proteome.fa # proteome.fa 蛋白序列,需要是fasta结构 # pfam_out.tbl 以表格形式输出的pfam比对的结果 # 比对的evalue值为最常用的默认参数2、提取Pfam蛋白预测结果整理成为excel表格的形式 按照我写的一个python脚本文件,可以将上面生成的 pfam_out.tbl 文件中的各个蛋白的Pfam结构域信息提取出来。 python脚本的下载地址如下:下载地址 python3 annotation_extraction_pfam.py改脚本会生成两个excel表格文件: prediction_info_from_pfam.xlsx:就是把pfam_out.tbl文件整理成为excel表格的形式。 proteome_annotation_pfam.xlsx:就是提取出来的每个蛋白的pfam结构域的注释信息,其中如果一个蛋白有多个结构域,不同结构域就会用“; "隔开。 如果脚本运行错误有可能是最新的Hmmer的版本不同导致输出结果不同导致的,可以自己修改python脚本或者下载比较早的hmmer用这个脚本。
上面用来比较的蛋白序列如下: >A0A067NEU8MAPLHLLAISLVWLLLLQTCTGRIYSKLSDLPTTRFDFVIIGSGAAGNVLANRLTENPHISVLVLEAGGSNEGVLDSEVPFFCPKLSPGTPFDWNFTTTPQAALNNHTVAFPRGFMLGGSTSINFLAYTRGSMDDWNRFAAITGDKGWSWHRLQRYFRKNEHFTRPADHHNIIGEFNPRVHSRRGINSVSLGGFPRPIDQMVLRAMQELPDEFPFNVDMNSGKQIGIGWTQGTIKGGRRSSSATSYLGSKFIRRRNLHVLLHARVTRLLQDGVTGGKKSFRKVEFAQNAPGSGVNGPFGPRRTILASKSVILSAGSVMSPNILMHSGIGDEQMLRRVGIKPLHNLPSVGQNLIDHSLTRLAWNVNSTDTYETHLRNATIAAADLAEWKRSETGYLTTTRFSFLGWLRLPKNATIFQRFKDPAAGLDTAHIEMLIGVSSAFTNGMNLAPPTGNFLSLSVGVVSPASRGSITINSSNPFDPPLIDPGLMTSEFDLFTMREAIRSVARFLSAPAWAGYILSPAGALADVDLSSDEQLNAYIRNNTGSLFHPVGTSSMSRRGSSSGVVDPDLKVKGIEGVHVVDASVMPRVPAAHTQVPTYVVAERAADLIKEAWDCY |
【本文地址】
今日新闻 |
推荐新闻 |