分享:听老师讲讲HPLC的计算 |
您所在的位置:网站首页 › 根据峰面积计算含量 › 分享:听老师讲讲HPLC的计算 |
分享:听老师讲讲HPLC的计算
|
我们看到,其流动相是:甲醇-0.2mol/L磷酸二氢钠溶液(pH=2.7)(42:58 v/v),那么也就是说有水相识0.2mol/L磷酸二氢钠溶液,其pH=2.7(用磷酸调节),有机相是甲醇。
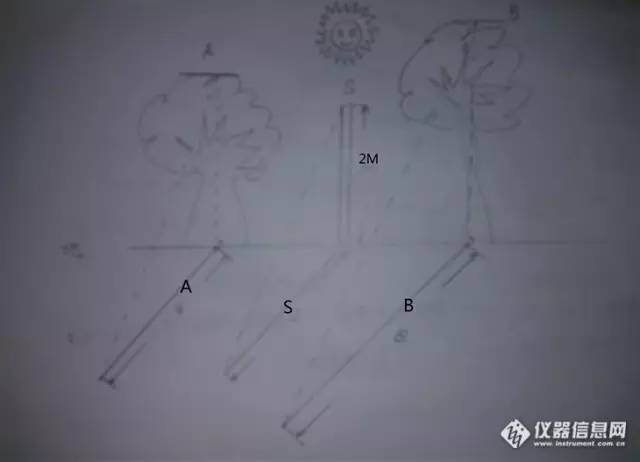
要注意,对于HPLC用的试剂,水,溶剂,都是需要好的。试剂一般在分析纯以上。最好用优级纯,溶剂一般用HPLC级别。水用超纯水。所有的溶剂,溶液,都需要用0.45微米(或更细)的微孔滤膜滤过。也要保证溶剂瓶清洗干净。干燥。 流动相脱气是很重要的,现在一般的仪器都有在线脱气。当然,在配制流动相前脱气,亦重要。一般脱气又抽真空脱气,超声波脱气,吹He脱气,加热回流脱气等。最常用的是前两个脱气方法。当然,抽真空脱气效率比较高,但是要注意有机相是否在抽真空后比例改变的问题。所以,对于混合两相的流动相,最好单独滤过,再混合。混合后一般会产生气体,用效率略低的超声波脱气即可。超声波脱气,只要看到无明显气泡即可使用。 “ 二.含量计算 对于HPLC,我们一般使用外标法计算。那么有同学问,那么为什么内标法很少在HPLC上使用呢? 那么我们看下气相色谱和HPLC不同的进样方式。GC手动进样还是依靠进样针的定量进样,因为进样量很少,以微升计,所以,稍稍不留神,就有可能多进或者少进样。而且,进样针上的刻度也很粗,所以,GC用手动进样,使用外标法确实很考验测试者的娴熟技巧和过人的眼力。而内标法在GC进样测试中,由于内标物的调节作用,可以使得由于进样误差降低到最小,其相关的文章,查阅qinqingcao在本论坛的文章。但是HPLC进样是六通阀进样,其有一根定量环,如果手动进样能够保证定量环是充满样品溶液的,那么进样误差就几乎不存在。况且内标法需要寻找合适的内标物质,同时配制内标物,比较麻烦,所以在HPLC中很少听说用内标法定量计算。更多的,是使用外标法计算。 所以,HPLC手动进样一般使用3倍量的定量环进样。也就是说,20微升的定量环,我要注入60微升以上的样品溶液。保证定量环中充满着样品溶液。而多余的样品溶液会被排除。这样保证每次进样都是定量环中的那些量。对于定量环,这个体积一般也不是正好是20.00微升,但是我们保证进样过量(多余的溶液会被排出),则能够保证进样都是按照定量环中的体积进入色谱系统,这样消除进样的误差。对于定量环,在做完样品后需要用无溶质的流动相清洗数遍。如果定量环真的很脏,无法清洗了,那么可以裁剪一段管径和定量环相同的干净的不锈钢管,裁剪长度和定量管相同,安装上去。当然资金充裕的话,也可以购买厂家的定量环。 当然现在自动进样器很普及,那么使用自动进样器就更加方便了。 对于外标法,实际上也就是用已知浓度的对照品(标准品)来对样品中的该物质。这实际上很象砝码和天平。标准品就如同砝码,在同样的色谱条件下,标准品的峰面积和浓度已知,而样品中的峰面积可以测试出来,这样通过比例式子比较出样品中该物质的浓度了。 在这里,我想和大家说明的是,一般是通过保留时间做定性的。比方说,我测试维生素C,维生素C标准品在一定的色谱条件下,其保留时间为2.4分钟;样品在同样的色谱条件下,2.4分钟有峰,一般认为是样品中也许有维生素C。当然,保留时间定性确实有其局限,但是没有其他手段,那么保留时间还是能够发挥定性的作用。 对于定量,外标法定量,如果做的准确的话,需要用多点(浓度)定量。然而在企业用单点法定量还是很多的。对于做线性,精密度,回收率,这些内容我们在方法学验证中讲授,这里我还是着重讲解单点法校正。 在讲单点法校正,我先问同学们一个问题,如何用简单的方法来测试校园中的树高度?
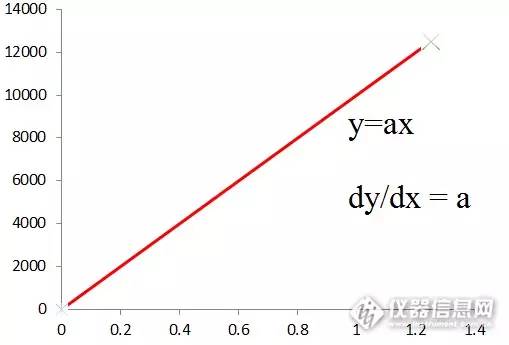
通过一根已知高度的标杆,在太阳下投射在地面的阴影的长度,计算出树的高度。同样道理,我们也是用已知的标准品的浓度和峰面积,用样品中同一物质的面积,计算出样品中的那个物质的浓度。根据稀释倍数计算出样品中,该物质的含量。 如果与标准曲线联系起来,这个就是单点法。也就是说,以0为圆心,标准品浓度和面积在二元坐标的一点的连线作为校准曲线。得y=ax 其中的a就是斜率,也就是面积随着浓度的变化而变化,其变化率为a。
那么样品浓度的计算,就是按照其峰面积在坐标上的点,对应的浓度。用以下公式计算。 单点法:
我们给出一个例题。 测试果汁中维生素C含量。配制20mg/100ml的维生素C作为标准品。称取20.005g果汁到50ml容量瓶中,加入流动相溶解定容摇匀。滤纸过滤。吸取滤液10ml到50ml容量瓶中。流动相定容摇匀。标准品和样品均吸取20μl进色谱,分别得到峰面积24336(标准品),20129(样品)。计算果汁中维生素C含量(mg/100g)
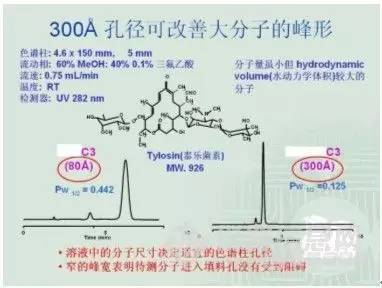
“ 三、总结 今天qingqingcao讲了HPLC中的计算,首先是流动相的计算,同时简单讲了流动相配制的要点。其次讲了含量的测试,着重讲了单点校正之外标法。补充了内标法不太用于HPLC的大致原因,给出了单点法校正的计算例题一枚。希望大家能够掌握这简单的计算,下次我们讲讲方法学验证的内容。其中要讲授外标法之多点校正,精密度,回收率等。 液相色谱方法开发的一般规则 高效液相色谱方法的开发是一个繁复的过程,但不管再繁复,也有其规律可寻。方法开发过程中,一个总的原则是:先找到目标化合物的峰,然后调整峰形,再是进一步完善。 1. 找到目标化合物的峰: 要找到目标化合物的峰,我们该如何开展工作,先举一个例子,下面是分析氟康唑氯化钠注射液的一个谱图: 谱图上的内容主要包括:色谱柱、波长、流动相、温度、流速和进样量这几项。这意味着如果这几个色谱条件都确定下来了就可以基本认为这个方法已经开发成功,所以开发方法时我们可以通过逐一的考查这几个色谱条件来进行。 考查色谱条件是需要通过在色谱仪上进样来进行的,这就需要我们首先确立一个初始条件,即回答从何开始的问题。在回答好“从何开始”这个问题之前,我们先要了解我们的被分析物,就像一场战争,首先要知道自己的敌人是谁一样,我们要了解它的物理、化学性质,特别是化学结构式非常关键。所以,色谱方法开发工作可以通过如下的步骤来展开: 1)收集资料: 对它的分子量、结构式,以及在水、甲醇、乙腈、四氢呋喃、正己烷和异丙醇中的溶解度有一个初步的了解。对分子量的了解在选择色谱柱的过程中是非常重要的,因为色谱柱填料的孔径对化合物的分离具有重要的影响,例子如图:
由此可以看出填料孔径对分离度和峰形是有一定影响的,120A的色谱柱通常适用的范围为分子量 |

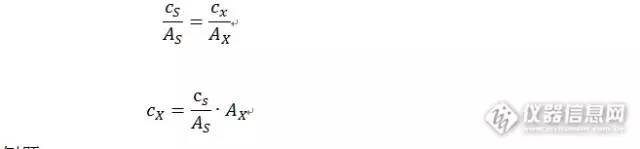


【本文地址】
今日新闻 |
推荐新闻 |