如何以Excel为基础做RNA |
您所在的位置:网站首页 › 有两列数据如何做成Xy散点图含名称 › 如何以Excel为基础做RNA |
如何以Excel为基础做RNA
|
B: Venn Diagram (韦恩图) C: Intensity Scatter Plot (密度分散图) D: Heatmap (热图) E: Gene Ontology (GO) analysis F: GO analysis 如果你认真看完本篇文章,你也能够做出从A到F这些图。 结合之前文章提到的配色方案(关于配色问题,参考以下链接),你会做出很漂亮的A-F,然后开心地放在PPT和论文中! 漆黑的师兄:为什么 PNAS 的图画得往往没有《Nature》、《Science》精致?
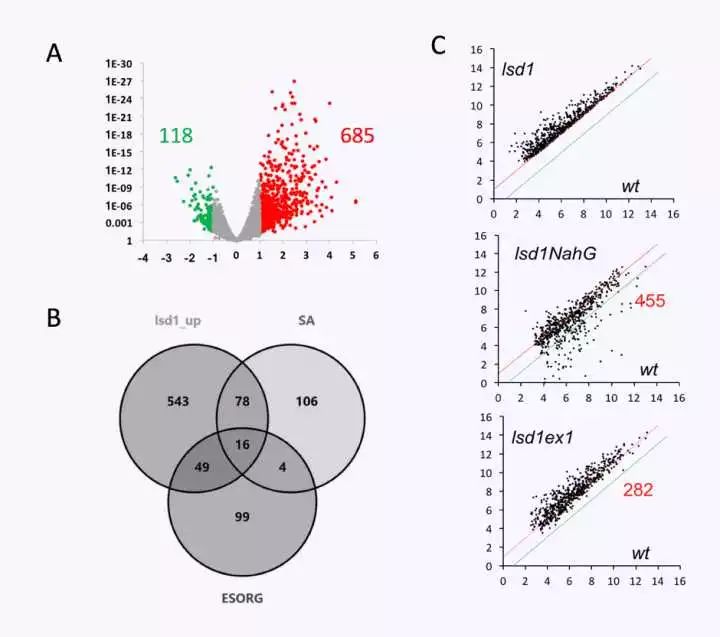
A: 火山图 B: 韦恩图 C: 密度散点图
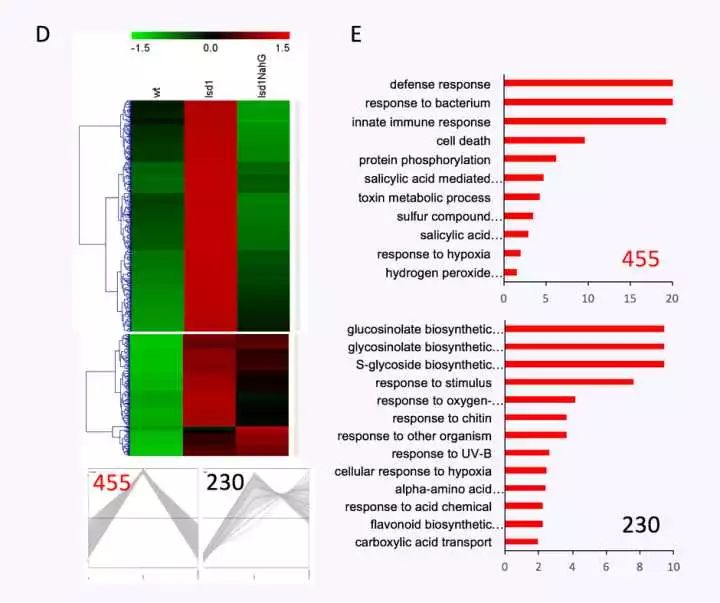
D:热图 E:GO分析
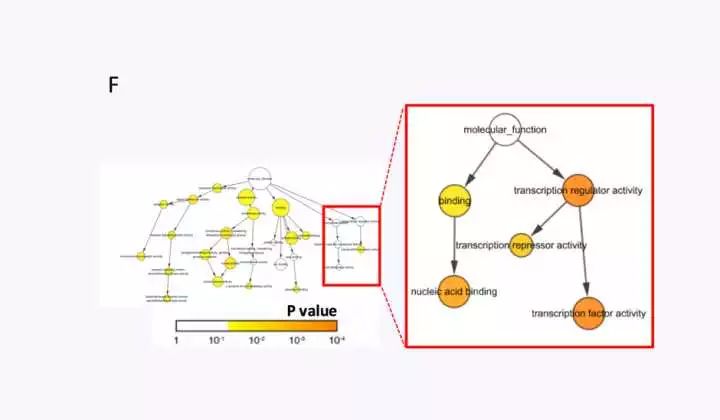
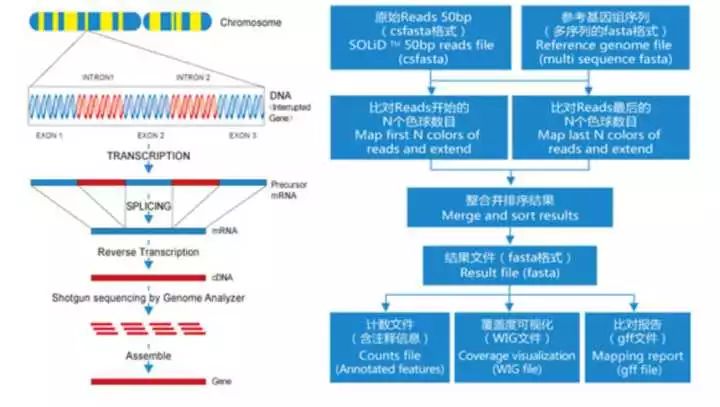
F: GO 分析 BiNGO RNA-seq的一般流程是这样的
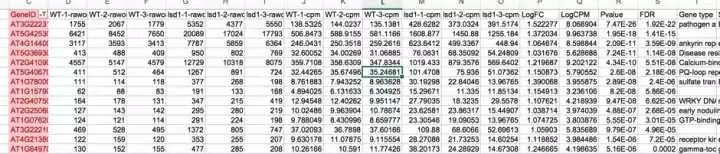
RNA-seq 流程图,图源网络,侵删 mRNA-seq 测序的最原始结果是核苷酸序列,文件大小在4-10GB之间,分析起来相当的繁琐,分析也不是一般的小本本能跑起来的,好在一般来说,我们从测序公司拿到的数据,已经是经过比对和Mapping之后的结果,公司给这个样子的表格:
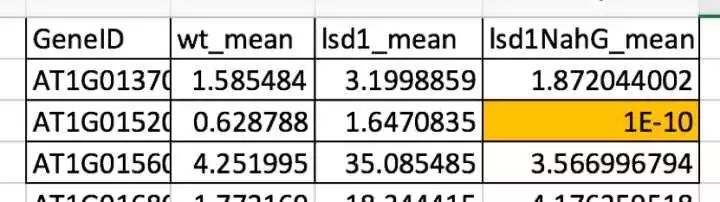
从公司拿到的excel表格 做好分析,首先要理解每一列每一行代表的意思,那么每一列代表的是什么意思呢? 1) GeneID: 分析出来的基因在基因数据库中的名称。 2) 从第二列到第七列, 分别是WT-raw-counts(-1,-2,-3)和lsd1-counts(-1,-2,-3),可以简单的理解为改Gene在这次测序中读到的总次数。 3) 从第八列到第十三列,分别是WT-CPM(-1,-2,-3)和lsd1-CPM(-1,-2,-3),可以简单的理解成一个是对照组WT另一个是实验组(Var2),CPM( counts per million reads)每一百万次读数该基因记到的次数。 4) LogFC (FC: Fold changes),一般是以2为底数的对数值,代表这个基因在实验组的表达是对照组的多少倍。LogFC≥1(upregulated上调)或≤-1(downregulated下调),即表达量大于等于2或者小于等于2,认为是比较显著的,是很重要的一个参数。 5) LogCPM (CPM, counts per million reads)一般好像用不到这个值,不知道为什么。 6) P_value(基因表达差异的可信度)一般取小于0.05。 7) FDR(False discovering rate)也是指示基因表达差异性是否可靠的值,一般取小于0.05。 8) Gene Type,这是根据数据库对基因的注释(Gene Annotation),一个基因可以从三个方面进行阐释,1)Biological Process; 2) Molecular Function; 3) Cellular Component,即这个基因参与到的生物进程(信号通路),表达的蛋白有什么样的分子功能,表达的蛋白亚细胞定位在哪里,三个方面。 1) GeneID: 分析出来的基因在基因数据库中的名称。 2) 从第二列到第七列, 分别是WT-raw-counts(-1,-2,-3)和lsd1-counts(-1,-2,-3),可以简单的理解为改Gene在这次测序中读到的总次数。 3) 从第八列到第十三列,分别是WT-CPM(-1,-2,-3)和lsd1-CPM(-1,-2,-3),可以简单的理解成一个是对照组WT另一个是实验组(Var2),CPM( counts per million reads)每一百万次读数该基因记到的次数。 4) LogFC (FC: Fold changes),一般是以2为底数的对数值,代表这个基因在实验组的表达是对照组的多少倍。LogFC≥1(upregulated上调)或≤-1(downregulated下调),即表达量大于等于2或者小于等于2,认为是比较显著的,是很重要的一个参数。 5) LogCPM (CPM, counts per million reads)一般好像用不到这个值,不知道为什么。 6) P_value(基因表达差异的可信度)一般取小于0.05。 7) FDR(False discovering rate)也是指示基因表达差异性是否可靠的值,一般取小于0.05。 8) Gene Type,这是根据数据库对基因的注释(Gene Annotation),一个基因可以从三个方面进行阐释,1)Biological Process; 2) Molecular Function; 3) Cellular Component,即这个基因参与到的生物进程(信号通路),表达的蛋白有什么样的分子功能,表达的蛋白亚细胞定位在哪里,三个方面。 当初我拿到这个含有上千甚至上万个基因的Excel表格是,一阵莫名激动,感觉自己马上就要发文章成为大佬受万人膜拜,前途亮的能瞎眼。然而,一股莫名的忧伤涌现,那么多基因,我该怎么办呢?总不能把基因都贴满论文吧?好像没有这样搞的啊! 后来才知道,第一步,先什么都不管,直接把所有基因进行可视化处理,就是上文提到的图A,火山图; 图D, 热图; 根据情况用C。 Volcano Plot 绘制起来比较简单,用到的值就是上文EXCEL表格中的P_value和LogFC。如果要绘制成像图A,灰色的是不显著的基因,红色的是显著上调的基因,就需要在EXCEL里的根据logFC 和P_value,把基因们分为三类(这点,请熟练运用EXCEL中的筛选,和排序功能) 1)P_value大于0.05,不显著的, 2)P_value小于0.05, LogFC大于等于1,显著上调的, 3)P-_value小于0.05, logFC小于等于-1,显著下调的。 1)P_value大于0.05,不显著的, 2)P_value小于0.05, LogFC大于等于1,显著上调的, 3)P-_value小于0.05, logFC小于等于-1,显著下调的。
Excel 中的排序和筛选功能
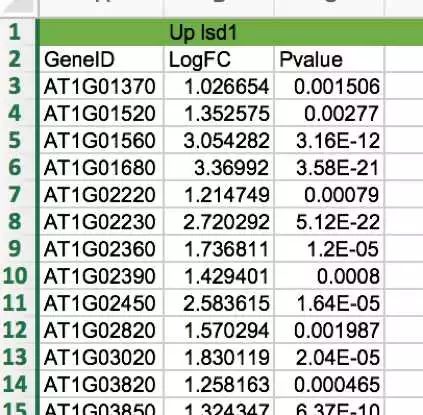
按P_value 和LogFC 分类后的数据 **选中以上三组数据中的任意一组,比如1)P_value大于0.05,不显著的, LogFC和P_value两列,X轴设置为LogFC, Y轴设置为P_value 点击 插入图标>选择XY 散点图> 是不是得到一个很奇怪的图,没关系, a) 可以点击Y轴,设置Y轴格式,选中 “对数刻度”和“逆序刻度值” b) 点击X轴,设置X轴格式,选择“标签”>“高”, c) 将纵坐标交叉,自定义设置成最小值 d) 选中图中的点,设置一下数据系列格式,找到“标记”,设置一下标记的大小和颜色(文中设置成灰色) e) 右击散点图,选择“添加数据”分别把其他两组数据添加进去,Volcano Plot就做好了。 a) 可以点击Y轴,设置Y轴格式,选中 “对数刻度”和“逆序刻度值” b) 点击X轴,设置X轴格式,选择“标签”>“高”, c) 将纵坐标交叉,自定义设置成最小值 d) 选中图中的点,设置一下数据系列格式,找到“标记”,设置一下标记的大小和颜色(文中设置成灰色) e) 右击散点图,选择“添加数据”分别把其他两组数据添加进去,Volcano Plot就做好了。
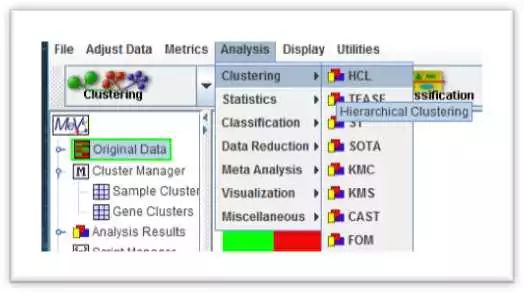
插入XY散点图 Heatmap 绘制起来要稍微麻烦一些,我暂时还没找到可以用Excel绘制Heatmap比较简单的方法或者程序包。为了简便起见,我选择了一个免费软件MeV (Multiple experiment viewer) 下载地址:http://www.mybiosoftware.com/mev-4-6-2-multiple-experiment-viewer.html 这个软件功能很强大,也可以用作Microarray data analysis,有包括HCL,K-mean Clustering 在内的好几种聚类分析方案,重要的是一键可用。好用虽然好用,但是前期的数据准备还是要在EXCEL里面先做好,这次用的是CPM,即上文提到的WT-CPM(-1,-2,-3)和lsd1-CPM(-1,-2,-3)。 a) 首先,算一下均值, “=mean(wt_cpm-1, wt_cpm-2,wt-cpm-3)”和“=mean(lsd1_cpm-1, lsd1_cpm-2, lsd1-cpm-3)”。如果,你的数据很多,比如在图D中,还有一个lsd1NahG,也可以算好均值,但是要注意的是,将每一个基因一一对应好,不要有错漏,有些基因可能在一个样本里没有检测到,而在另一个样本里有。那么基因样本的缺省值,可设置0.00000001,意即表达量极低,以至于不能检测到。也可以设置能NA,意为错误或缺省值。两种处理方式,在文章中都是可以接受的,但为了美观,我通常会选择前者。后者,用MeV作图时会出现一块灰色的区域。 a) 首先,算一下均值, “=mean(wt_cpm-1, wt_cpm-2,wt-cpm-3)”和“=mean(lsd1_cpm-1, lsd1_cpm-2, lsd1-cpm-3)”。如果,你的数据很多,比如在图D中,还有一个lsd1NahG,也可以算好均值,但是要注意的是,将每一个基因一一对应好,不要有错漏,有些基因可能在一个样本里没有检测到,而在另一个样本里有。那么基因样本的缺省值,可设置0.00000001,意即表达量极低,以至于不能检测到。也可以设置能NA,意为错误或缺省值。两种处理方式,在文章中都是可以接受的,但为了美观,我通常会选择前者。后者,用MeV作图时会出现一块灰色的区域。
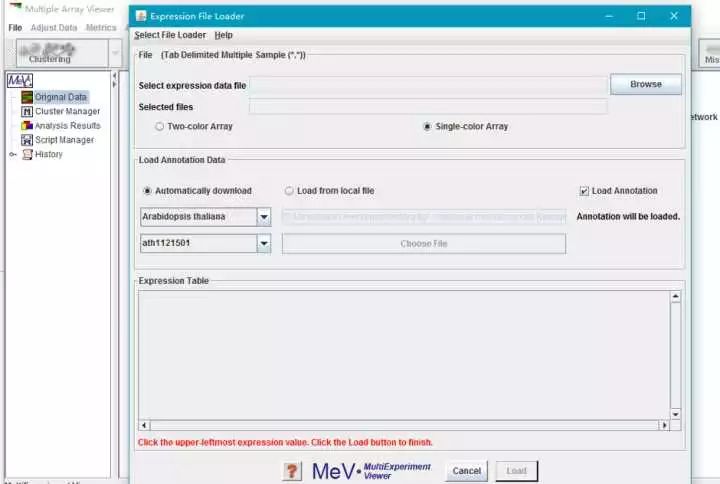
计算每个样本的均值 b) 接着,把整理好的数据,复制到txt文档中,因为MeV只支持txt格式的文本文档数据。 c) 打开软件,File>load data ,注意软件左下方的红字:Click the upper-leftmost expression value. Click the load button to finish. b) 接着,把整理好的数据,复制到txt文档中,因为MeV只支持txt格式的文本文档数据。 c) 打开软件,File>load data ,注意软件左下方的红字:Click the upper-leftmost expression value. Click the load button to finish.
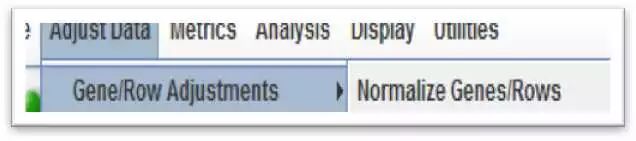
MeV软件界面,导入数据 d) 依次点击Adjust data 选择 e) d) 依次点击Adjust data 选择 e)
调整数据,对数据进行标准化 f) f)
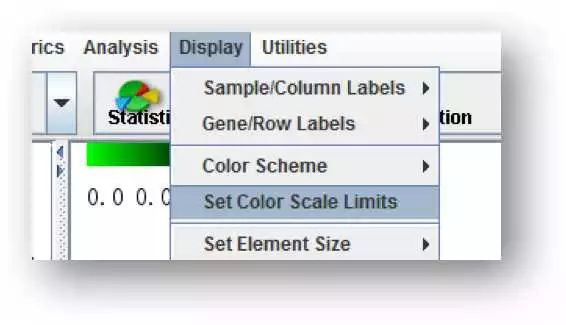
设置颜色限制范围 g) g)
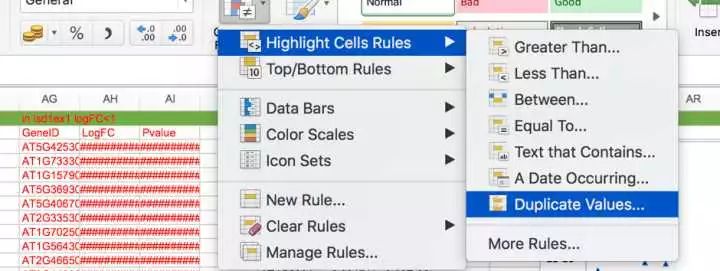
对基因进行聚类分析 HCL clustering可以用软件的默认设置,具体每一条什么意思,大家可以查询一下,解释起来很麻烦,而且也不是本文的重点。如果样品不是很多可以不用选Sample Tree. 具体的软件使用,可以参考软件使用手册,一定要读一遍手册!!!然后调整一下字体什么的,就做成图D了。 韦恩图 的意思一般是,找到两个或者三个样本之间相同的差异表达基因,比如与wt比较,样本lsd1中有很多LogFC>1的基因,lsd1NahG也有很多LogFC>1的基因,那么这些基因中有没有重叠的(Overlapped Genes),这时就可以用韦恩图了。这时,用的就是GeneID,可以 a) 同时选中两列GeneID,用EXCEL中的“条件格式”>“突出显示单元格规则”“重复值”把重复值标记成红色选项,手动作图。
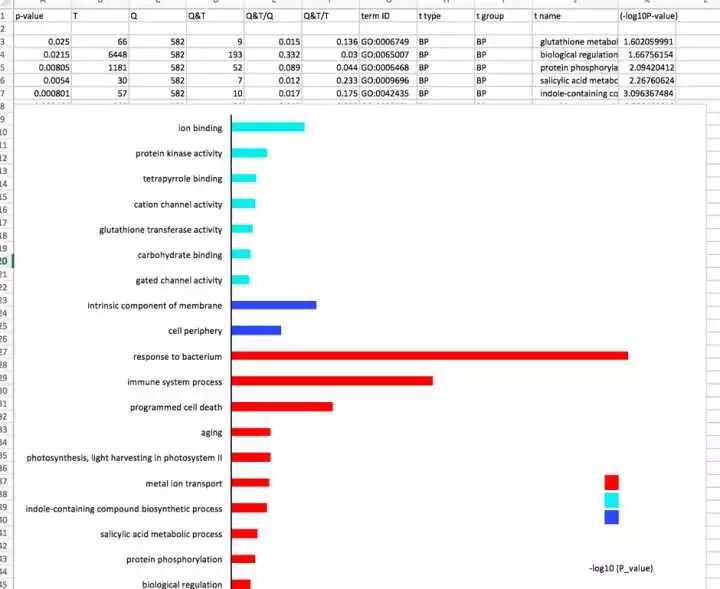
利用Excel中的条件格式功能做韦恩图 b) 也可以用现成的工具 http://bioinfogp.cnb.csic.es/tools/venny/ 这样 图B就做好了。 GO analysis 现在,我们已经把这些基因分类了,也Visualize了,其实这些炫酷的图基本没啥卵用的。也就是为了好看,告诉读者,我做了RNA-seq结果是这样的,给你看给你看,多好看啊!然而,读者肯定会一头雾水!这是个啥,啥,啥!!!!!为了解释这是个啥啥啥!我们就要对这些基因进行功能性分析,也就是Gene Ontology(基因本体论)从,三个方面 1) Biological Process (BP); 2) Molecular Functionv(MF); 3) Cellular Component (CC), 1) Biological Process (BP); 2) Molecular Functionv(MF); 3) Cellular Component (CC), 对这些基因进行注释,然后找到一些线索,为接下来的研究、设计实验做些准备。一些人开发了Excel分析包,我没用过,因为用于GO analysis的分析软件特别多。一款比较常用的就是 gprofiler: http://biit.cs.ut.ee/gprofiler/ 这里用到的是GeneID 把得到的数据,以excel的方式存起来,打开文档把corrected p-value 用-Log10作一下转化得到-log10(p-value),简单的按从小到大排一下序,然后选中term name,插入条形图,就可以做出图E。按颜色,把BP,MF,CC标出来。
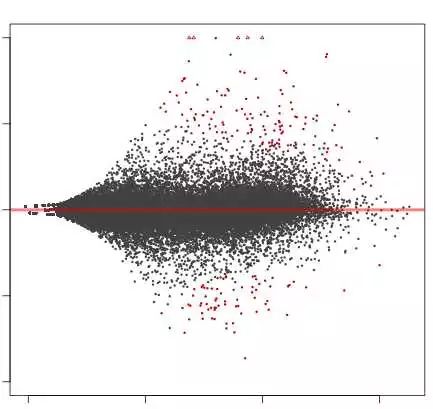
利用Excel做出条形图,可视化GO分析数据,不同颜色代表不同功能类型 红色:BP,深蓝色:CC, 浅蓝色:MF 图F表述的意思其实和图E差不多,根据自己的需要可以做出选择,作图F用到的也是GeneID,不同的是,它是用一个软件Cytoscape:http://www.cytoscape.org中的App BiNGO做的,操作起来很简单,有兴趣的可以去下载软件自己去试一试,这里就不详细说了。 最后,还差一个图C,图C是干嘛的呢?图C学名Ratio -intensity plot 假如,有一些基因在mutant lsd1 里面是上调的(图C中全部为红色),那么这些上调的基因,在敲出一个关键基因以后,表达量会不会受到影响呢?这时候就要用到图C:密度散点图(Intensity scatterplot),这里用到的值是“mean(wt_raw counts-1, wt_ raw counts -2,wt_raw counts -3)”和“mean(lsd1_raw counts -1, lsd1_ raw counts -2, lsd1_raw counts -3)” 首先,将两组数据的均值用log2做一下转换。然后选择转换后的数据,插入XY scatter,编辑一下X、Y轴就可以了。 还有一个类似的,叫做R-I (MA)plot,看起来像是把上述密度散点图旋转45°,也是一种比较初级的Normalization的方法,一般的用于全基因组基因可视化绘制,用的也是mean(wt_raw counts-1, wt_ raw counts -2,wt_raw counts -3)”和“mean(lsd1_ raw counts -1, lsd1_ raw counts -2, lsd1_raw counts -3)”,也做转换 不过不同的是: I=log2(wt_mean*lsd1_mean):作为X轴 R=log2(wt_mean/lsd1_mean):作为Y轴 大概是这个样子的。
R-I 或MA plot 好了,如果你认真看完这篇貌似技术贴的水贴,那么就可以解决你的燃眉之急,解决一大半类似的数据分析问题了。 我现在也在学习R,以及Python。因为,要真正掌握生物信息学的精髓,这些是远远不够的。虽然我只是一只分子植物学喵!如果你能熟练运用R语言,那么做出以上图,将会更加容易。 最后,欢迎大家提出各种意见,欢迎各种姿势骚扰,交流学习经验。 本文以获作者授权以原创发表 如需转载请联系EasyCharts团队! 微信后台回复“转载”即可!返回搜狐,查看更多 |
【本文地址】