Heliyon:新药研发,AI入场 |
您所在的位置:网站首页 › 新药开发过程包括 › Heliyon:新药研发,AI入场 |
Heliyon:新药研发,AI入场
|
原创 Cell Press CellPress细胞科学  交叉学科 Interdisciplinary 当前,新药研发面临着成本高企、收益率下降的双重困境。新药研发具有技术难度大、投入资金多、研发风险大、回报率高和研发周期长等特征,随着疾病复杂程度的提升,新药研发难度和成本迅速增加,全球新药研发成功率呈明显下降趋势。人工智能的发展,为新药研发带来了新的技术手段,大大提升了新药研发的效率,为降本增效提供了可能。AI技术作为提升新药研发效率的重要驱动力量,正在加速对新药研发各环节的渗透。本项研究对于AI在新药发现全过程中的参与进行了详尽地综述。2023年6月25日,这篇文章以“AI in drug discovery and its clinical relevance”为题,发表在Cell Press细胞出版社期刊Heliyon上。  论文摘要 新冠疫情使得新型药物发现成为热门。然而一种药物从构思到最终临床使用,需要经过一个漫长、复杂且昂贵的过程,存在着许多潜在的困难。过去十年里,计算硬件(云计算、GPU和TPU)发展进步,深度学习强势崛起,医学信息也随之快速增长。大规模分子筛选、个人健康或病理记录以及医学数据都可以通过人工智能(AI)进行分析,以加快新药研发进度,规避潜在困难。本文展示了包括计算方法的新药设计和药物性质预测在内的新药发现各个阶段应用AI的情况。探讨了开源数据库和基于AI的软件工具,以及它们所涉及的分子表示、数据收集、复杂性、标记和标签之间的差异等问题。还探讨了当代AI方法(如图神经网络、强化学习和生成模型)以及基于结构的方法(如分子动力学模拟和分子对接)如何有助于药物发现应用和药物反应分析。最后,还讨论了基于AI的生物技术、药物设计初创公司的最新发展和投资情况,以及它们目前的进展、期望和推广。 简介 全球每年有6%至7%(8.5至9万亿美元)的国内生产总值用于医疗保健。推出一种新药物的成本超过10亿美元,时间可能长达14年。全球范围内的药物研发成功率在所有治疗类别中都非常低,有97%的癌症药物在临床试验中以失败告终。随着医疗记录的数字化,临床试验、精准医学、药物发现和卫生政策都开始受益于数据驱动的方法。过去十年里,新型分析方法和计算进步彻底改变了药物发现。 本文讨论了药物设计中的数据表示和预测等关键问题,以及AI在这些问题上的优势。许多药物发现任务因为缺乏适合AI的基准数据集和标准化知识表示,难以形式化为机器学习问题。例如,药物可以用多种不同的格式表示,如SMILES字符串、扩展连接指纹(ECFP)和图形,蛋白质还可以表示为1D氨基酸、蛋白质序列和3D结构。另一个问题是标签资源较低和标签之间的差异难以为机器指定有意义的学习任务。本研究还讨论了在药物发现流程的不同阶段使用机器学习库、不同分子表示和图神经网络的作用。 药物发现流程通常包括如下图所示的几个阶段。在基于靶点的发现中,首先是从大量蛋白质(生物体的蛋白质组)中识别与疾病有关联证据的新型靶点。通过高通量筛选化合物库来识别可能相互作用的分子。化合物将针对有利的药物特性进行优化,经过临床前和临床试验,并在理想情况下获得FDA批准。药物发现流程的所有阶段都可以从AI中受益,例如,用于新合成分子设计的生成模型,用于优化分子属性的强化学习,用于预测药物疾病关联、药物重用和药物反应的图神经网络。自然语言处理,也可以通过挖掘科学文献来找到药物,并自动化FDA批准步骤。 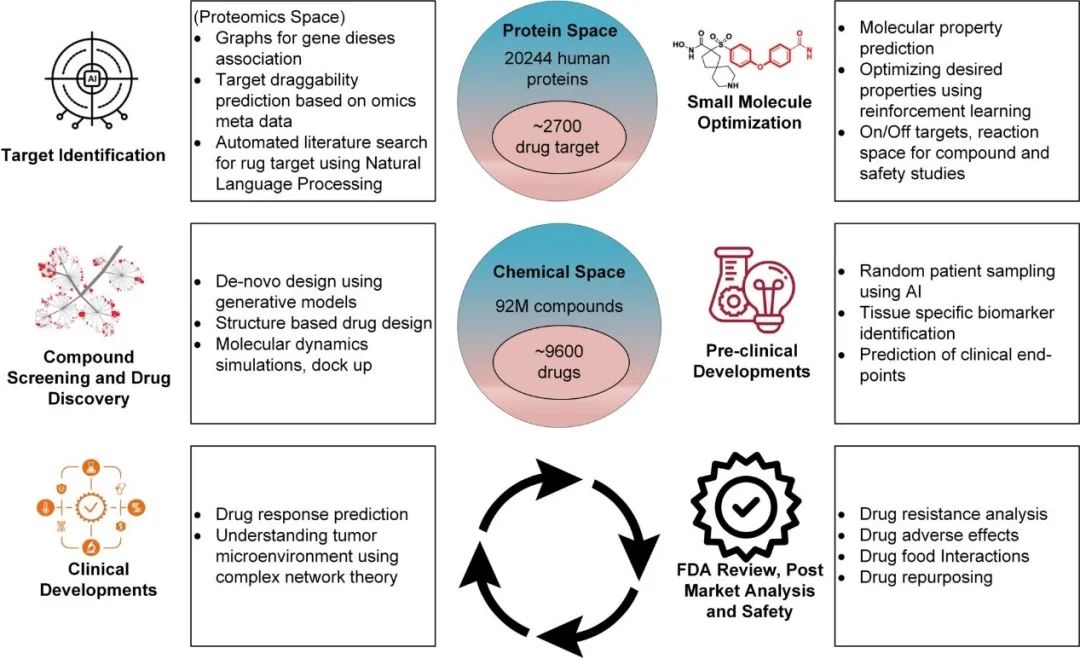 数据科学在药物发现过程中的应用 流感和COVID-19等流行病的出现,以及癌症和心脏疾病等严重疾病的流行,表明发现新药物仍然是人们的需求。新药发现通常是一个多阶段的过程,包括目标识别、验证、高通量筛选、动物实验、安全性和有效性协议、临床试验以及监管批准。开发一种新药物通常需要约14.6年,成本约为26亿美元。AI方法可以在该过程的多个阶段中应用,比如识别新型靶点,评估药物—靶点相互作用,研究疾病机制,小分子化合物的设计和优化。这些方法还可以用于识别和开发预后生物标志物,以及研究药物的疗效、反应和耐药性。 1.药物发现中的靶点识别 药物发现过程中的靶点识别旨在识别可能通过调节其活性来改变疾病状态的分子,通常是蛋白质。机器学习算法可以分析各种类型的数据,包括基因表达谱、蛋白质相互作用网络以及基因组和蛋白质组数据,以识别可能参与疾病途径的潜在靶点。在人类蛋白质组中约有2万个蛋白质,只有约3000个被认定为潜在治疗靶点。未来的知识可能会扩展我们对蛋白质成为药物靶点的理解。 确定靶点的第一步是建立靶点与疾病之间的因果关系。使用图形、图神经网络或基于决策树的方法可以识别基因与疾病之间的因果关系。有关靶点与疾病关联的主要信息来源是文献。文本挖掘和自然语言处理方法也可用于从文献中识别相关的靶点—疾病对,并开发用于靶点识别的数据库。BeFree、PKDE4J和其他基于深度学习的工具可用于挖掘文章,识别药物—疾病、基因—疾病和靶点—药物的关联。 2.化合物的虚拟筛选和优化 AI可用于虚拟筛选和优化化合物,估计其生物活性,并预测蛋白质-药物相互作用。AI在虚拟筛选方面的一种方式是通过开发预测模型,识别与目标蛋白质结合概率较高的化合物。这些模型可以使用各种类型的数据进行训练。药物的物理化学性质也可能会间接影响药物与靶受体家族的相互作用,因此在设计新药物时必须予以考虑。AI还可用于规划化学合成的有效路径,并深入研究药物的反应机制,以识别与其他分子的潜在不良相互作用。 3.临床前和临床研究 预测药物的可能反应是药物设计流程中的关键一步。相似性或基于特征的机器学习方法可以通过结合亲和力或自由能的结合来预测药物对个体细胞的反应,以及药物—靶点相互作用的疗效。相似性方法假设相似的药物作用于相似的靶点,而基于特征的方法找到药物和靶点的个体特征,并将药物—靶点特征向量提供给分类器。 AI技术可帮助通过识别相关的人体疾病生物标志物,并预测潜在的毒性或不必要的副作用。通过筛选高维临床变量来选择一组患者,从而为临床前试验选择潜在的患者。AI还可帮助在实际试验之前预测临床试验的结果,从而最大程度地减少对患者的任何有害影响。 4.FDA批准和后市场分析 自然语言处理(NLP)可用于挖掘科学文献,报告药物的不良反应,包括毒性、耐药性,并为监管批准或专利申请准备自动化评估。基于NLP的情感分析方法可用于推荐药物。基于机器学习的系统预测产品的可能销售量,有助于制药公司优化其商业资源。 现有的药物开发数据库和工具 1.化学和生物数据库 实验生物测定和计算产生的药物-靶点相互作用(DTI)数据需要整理在公开的数据库中。化合物和生物活性数据库列于表1中,而靶点和化学数据库则列于表2中。   2.基于人工智能的药物开发软件工具 AI工具有可能通过使研究人员能够快速分析大规模数据集、设计新分子并预测潜在药物候选物的功效,从而改变药物发现。  药物发现应用中的数据表示和图神经网络 分子的机器可读表示允许在药物发现的机器学习算法中快速计算、查询和存储分子,其质量可能影响数据的变异性的利用。 大多数机器学习算法假设训练和测试数据都是独立和同分布的,然而,这个假设对于药物发现应用并不成立。小分子优化和设计需要探索来自特定化学空间的结构变化。模型必须在分布偏移的情况下泛化以便有用。尽管存在分布偏移,但化学信息学和药物化学仍将受益于所学习的特征。在这里,我们讨论了分子表示学习中的一些关键进展。 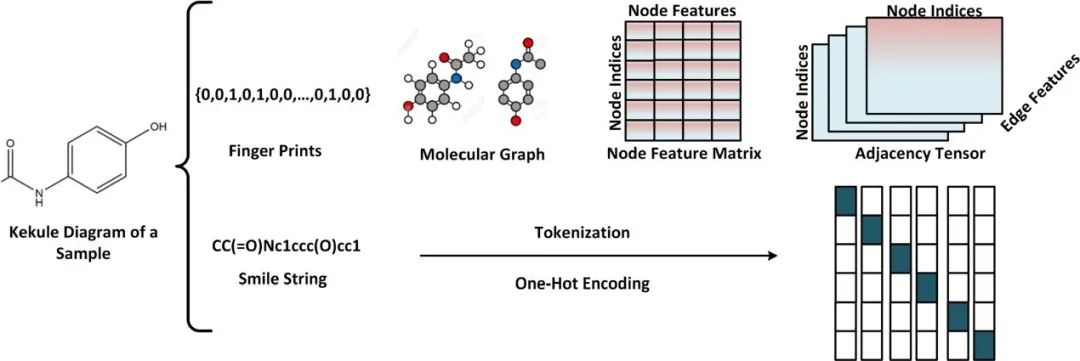 1.分子表示 固定的分子描述符可以根据其维度进行分类。分子具有0D特征,如分子量、原子数和原子类型计数。对于功能团,需要涉及更多结构信息的描述符,如指纹(二维二进制向量)。更复杂的表示,如SMILES、分子图和指纹,是为机器学习算法开发的(如上图所示:小分子表示的不同格式的图示。分子可以表示为带有键和原子的凯库勒图、SMILES 字符串,也可以表示为可以构建邻接、节点和特征矩阵的分子图)。机器学习模型中使用的分子描述符是固定的且不可学习的。 化合物和靶点可以在深度学习中用不同的编码表示;例如,可以使用Transformer编码器学习SMILES表示,使用循环神经网络学习蛋白质表示。分子还可以通过分子图(G =(V,E))直接嵌入到连续潜在空间中,无需进行特征工程,将原子或残基映射到节点(V),并将节点之间的化学键或连接分配给边(E)。 2.拓扑数据分析 拓扑数据分析(TDA)可以用于检查复杂的数据集,例如生物分子的表示。TDA基于代数拓扑学,它是数学的一个分支,研究通过连续变换保持的空间特征为TDA提供基础。 一种典型的预测化合物的蛋白质—配体结合亲和力的方法是将蛋白质—配体复合物可视化为一个持续图,一个几何对象。复合物的拓扑结构,包括连接组件的数量以及孔和空洞的存在,被描绘在持续图中。这种方法被称为“PerSpect ML”,即“持续谱基机器学习”,它在多个基准数据集上的蛋白质-配体结合亲和力预测优于现有最先进的方法。 3.图神经网络 大多数生物医学数据,如蛋白质相互作用、蛋白质—药物相互作用、药物—疾病相互作用以及药物重新利用等,都是相互关联的,因此适合用图来表示。小分子药物也可以用图来表示,其中原子作为节点,化学键作为边。知识图可用于展示药物、不良反应、药物重新利用和相关结果之间复杂的关系,以帮助产生新的假设。 图的一个重要结构属性是节点通常不需要以任何特定顺序呈现,并且在图上运行的函数应该是置换不变的(与顺序无关的),因此对于任意两个同构图,这些函数的输出应该相同。这个属性使得图成为表示分子和药物的合适候选者。分子图和子图可以轻松地映射到化学(亚)结构,使得它们易于解释。 图神经网络(GNN)是一种适用于药物发现的机器学习算法。GNN设计用于处理图数据,它表示实体(如化合物和蛋白质)之间的关系。GNN编码了节点之间的配对连接而不是非欧几里得空间中的点,捕捉了原子数据的结构化表示。典型的GNN由一个或多个层组成,通过递归地传递消息来从节点特征向量和相邻节点对进行置换不变的节点聚合,从而得到一个读取操作(如下图所示)。这个概念是图中的节点不断地与其邻居交换信息/消息,直到达到稳定的平衡。 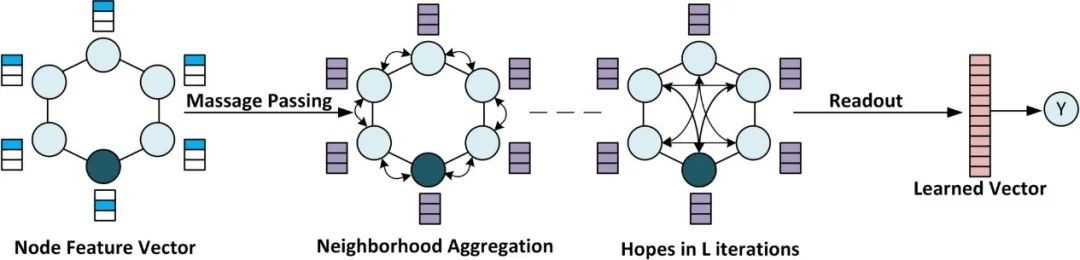 分子生成的深度学习模型 GNN同样可用于药物发现中的分子生成。基于GNN的模型可以通过学习给定数据集中原子和分子片段之间的关系来生成具有期望性质的新分子。在MolMP中,图的构建被建模为马尔科夫决策过程问题,其中图的开发、追加、连接或终止仅依赖于其当前状态,由神经网络控制采样过程 (如下图所示)。与基于SMILES的分子生成相比,MolMP在多个不同的评估指标上表现更优越。 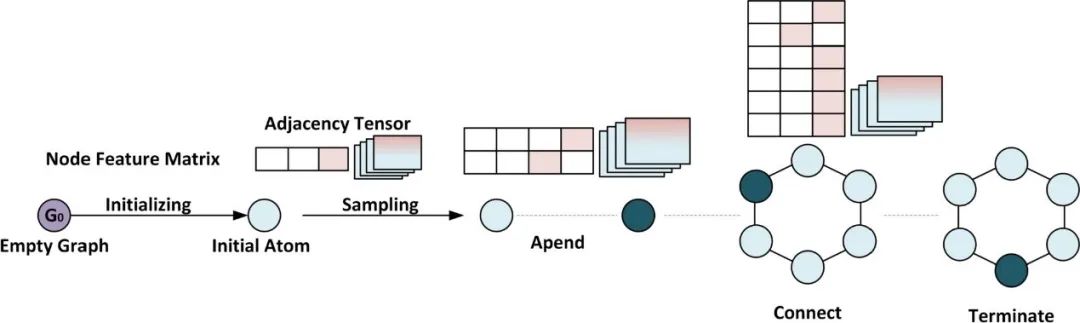 1.生成模型 深度分子生成模型能够快速探索大化学空间,包括通过合并现有化合物部分生成的新结构。通过使用遗传算法或粒子群优化,生成对抗网络能够生成具有期望性质的合成化合物或分子,并通过从学习的概率分布中进行采样来学习训练数据的概率分布并生成新的化学结构。化学指纹、SMILES、分子图、三维结构以及其他分子表示可以用于生成模型。然而,评估由生成模型产生的分子的独特性和最终相关性仍然是一个未解决的问题。 2.变分自动编码器 变分自动编码器(VAEs)被用于生成新的化学结构,这些结构通过无监督学习映射到了ZINC数据库。该模型由编码器、解码器和预测器组成。VAE将离散的分子结构转换为实值连续向量,解码器将其转换回离散结构。可以通过任何优化方法在连续潜在空间中搜索具有期望性质的新化学结构。 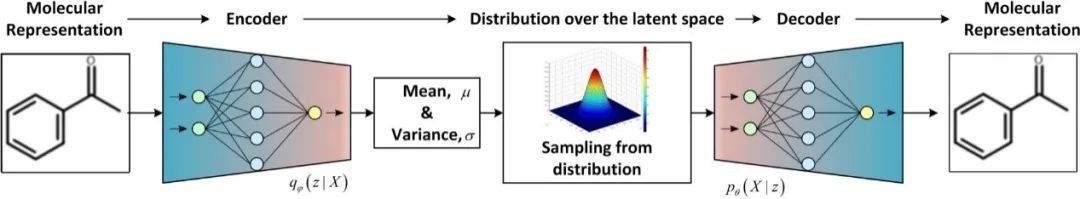 3.强化学习 使用连续数据驱动表示来控制生成分子的性质是困难的。强化学习(RL)是一种应用于药物发现中的分子生成机器学习算法。作为一种用于动态决策的机器学习范例,RL可用于设计具有最佳性质值(如溶解度、药代动力学性质或生物活性)的化学化合物。它涉及分析潜在行动,并估计这些行动与潜在后果之间的统计关系,然后确定一个旨在获得最佳可行结果的策略。深度RL试图从理论上无限的动作空间中找到最佳的行动集。这种算法的特性可以用于探索无限的化学搜索空间,避免了通过计算来检查每种可能的解决方案。 结构基药物设计 人类基因组计划的完成使得基因组学、蛋白质组学和结构数据爆炸式增长。由于生物信息学和数据分析方法的进步,优良的药物靶点正以更快的速度和低成本被识别出来。计算结构基药物设计充分利用了生物数据的积累,如蛋白质的结构(蛋白质数据银行)和药物数据库(药物银行)。关于潜在药物靶点的结构的知识极其宝贵,不仅适用于引物发现和优化,还适用于药物开发后期,出现毒性、药物耐药性或生物可利用性等问题时。如果对生物分子或复合物没有实验结构,可以使用分子建模软件来预测结构,其质量可以使用计算工具进行评估。 1.计算模型 尽管蛋白质数据银行和药物银行为大量蛋白质结构和药物复合物提供了高质量资源,但特定药物靶点复合物的结构信息可能无法获取,尤其是对于突变结构和药物-突变复合物。在这种情况下,可以使用计算建模来预测突变结构。Rosetta-Commons模拟蛋白质结构和大分子复合物。其他计算和统计方法可用于进一步评估预测模型的质量。 2.分子对接 分子对接用于预测分子在形成复合物时的相对方向,从而估计它们的结合亲和力。几种开源的分子对接软件包,如Auto-Dock、Flex-Aid和rDock,都是可用的。蛋白质是可移动的对象,它们的构象变化能力影响着分子对接旨在捕捉的蛋白质-药物相互作用。分子动力学模拟可用于预测蛋白质—药物复合物的时间依赖行为。 3.分子动力学模拟 分子动力学(MD)模拟了DNA、蛋白质和药物—靶点复合物等分子的运动(下图为执行 MD 模拟的流程)。它可用于识别蛋白质和复合物的自由能景观和生理构象,从而提供有关结构和蛋白质-药物复合物的生物活性的洞察。在MD模拟中,基于它们的位置、速度和加速度,所有原子的轨迹都可以使用牛顿第二定律获得。MD模拟计算成本高,需要有效的计算资源,如并行计算。MD模拟软件,如Amber、Gromacs和Charmm,提供了分析、可视化和预测蛋白质、药物和复合物性质的功能。分子动力学模拟计算可以利用机器学习技术加速。 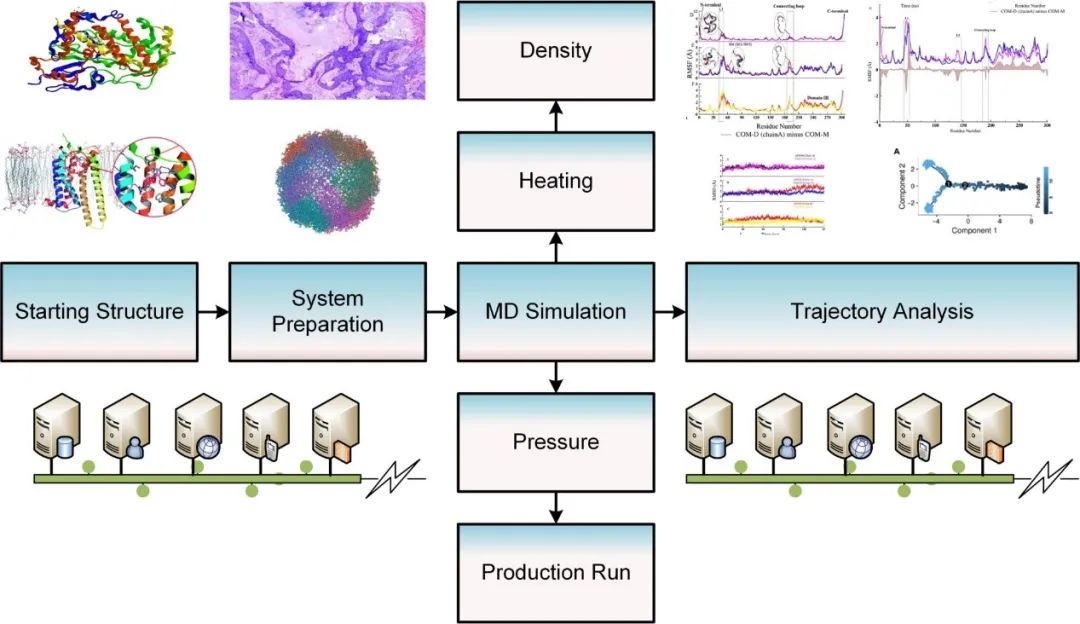 4.蛋白质-药物复合物的结合口袋建模 由于键的形成和蛋白质—配体相互作用而释放的能量称为结合的自由能,它可以用于估计结合亲和力并预测药物的响应。MD轨迹和Amber中的工具可用于计算结合的自由能。单个残基的能量贡献用于推断配体和蛋白质的结合模式。 下图显示了基于结构的对接和药物响应分析的框架。可以使用蛋白质数据银行、其他药物数据库或建模的目标结构来执行对接,然后进行MD模拟以研究构象、稳定性和结合自由能。现代几何深度学习方法可用于学习蛋白质-药物复合物的几何。药物或药物-剂量响应曲线显示了有机体或系统对药物随时间暴露的响应,常用参数是IC50,它衡量了物质抑制特定生物或生化功能的效力。IC50值通过昂贵的生物实验确定,并且容易出现错误。基于深度学习的方法可用于预测IC50值。 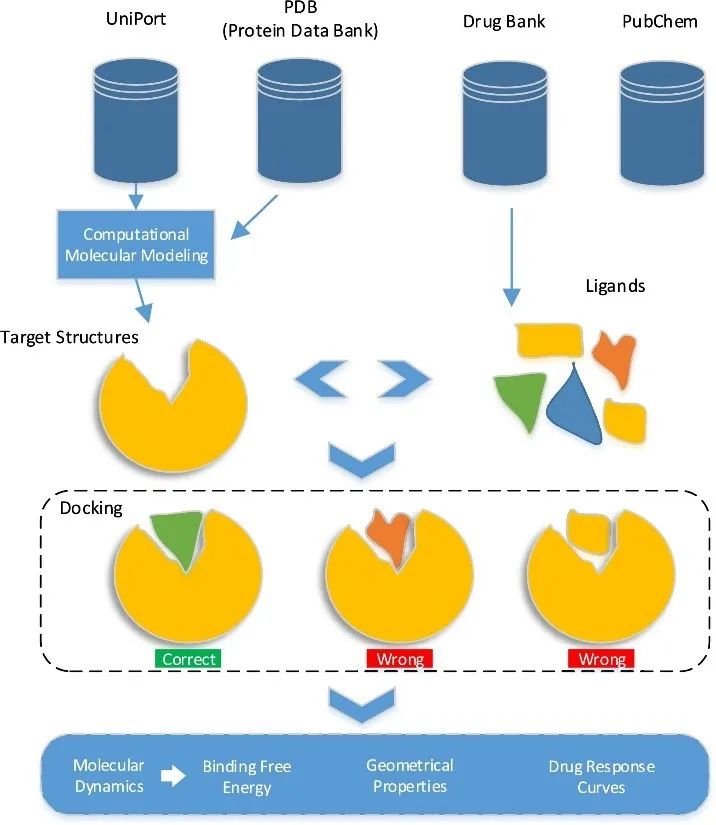 基于人工智能的制药初创公司 根据Emersion Insights的研究,2021年上半年,药物研发领域的人工智能初创公司筹集了约21亿美元的资金。大型生物制药公司在药物研发的各个阶段已经开始使用人工智能。辉瑞正在使用IBM Watson,一种基于机器学习的系统来搜索免疫肿瘤药物。罗氏基因泰克正在使用来自剑桥的GNS Healthcare,诺华正在使用微软进行细胞和图像分割研究,而阿斯利康与BenovalentAI合作,共同开发并商业化Jenssen的新型临床候选药物。谷歌、DeepMind、Insilico Medicine、Deep Genomics、Healx等公司也在人工智能药物研发应用上进行大规模投资。 美国是人工智能实施的先驱,也是全球药物研发领域中超过一半AI公司的主要参与者。近年来,美国和欧盟的投资者数量大幅增加。因此,这些地区以及英国在人工智能药物研发应用的投资者数量方面处于领先地位。诺华是英国和欧盟药物AI竞赛的重要参与者。两家总部位于英国的公司,BenevolentAI和阿斯利康,正在共同开发由AI生成的新型慢性肾脏疾病靶点。最近,中国也开始关注在药物研发领域的人工智能投资,并承诺投资50亿美元。中国最大的城市之一,天津,将投资160亿美元用于人工智能业务,而北京将创建一个价值21.2亿美元的人工智能发展项目。到2030年,中国计划成为基于人工智能的药物研发初创公司的领导者。 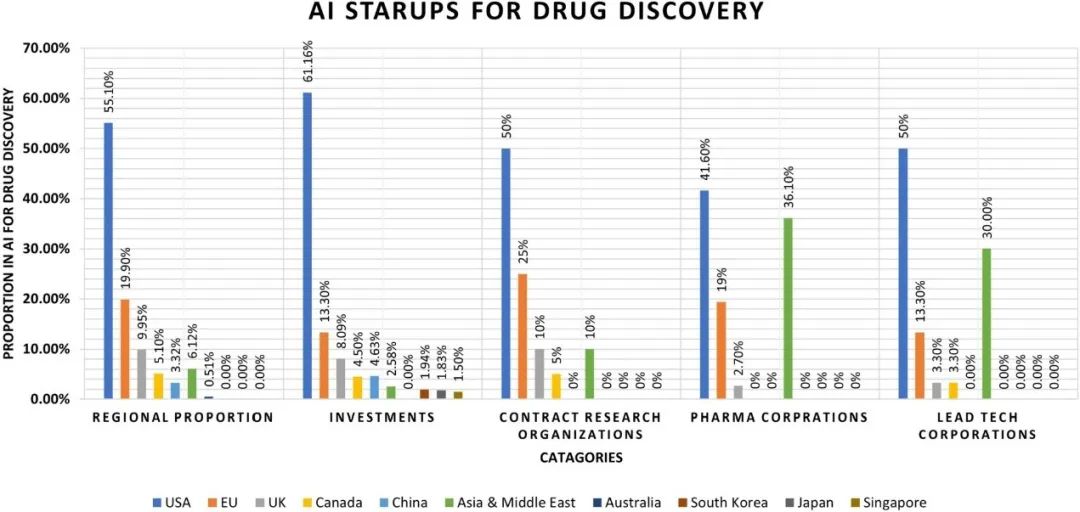 在合同研究组织(CRO)的人工智能竞赛中,美国处于领先位置,有50%的CRO位于美国,其次是占25%的欧洲。同时,亚洲也有10%的CRO专注于以人工智能为导向的药物研发。根据使用人工智能进行医疗保健和药物研究的IT公司数量,美国领先于所有国家。然而,在化学公司数量方面,亚洲排名第二,欧盟排名第三。考虑到欧盟近年来化学领域的增长,这在化学化合物和相关产品市场上超过了美国和亚洲市场,这是合理的。 结论 在药物设计、临床决策等领域,低成本、智能灵活的人工智能方法正在医疗保健行业内应用地如火如荼。药物设计道阻且长,但数据科学方法可以加速这一过程,以进行靶标识别、全新分子设计、药物再用、逆向合成、反应性和生物活性预测、FDA批准以及后市场分析等环节,一些制药公司也已经开始让人工智能入场。深度神经网络可以提高小分子性质推断预测能力,如果没有大量实验数据,也可以使用一次性学习。理解与底层数据相关联的技术和人为错误、标签约束以及生物变异是预测模型的关键。 * 参考文献见原文 相关论文信息 论文原文刊载于CellPress细胞出版社旗下期刊Heliyon上,点击“阅读原文”查看论文 ▌论文标题: AI in drug discovery and its clinical relevance ▌论文作者: Rizwan Qureshi, Muhammad Irfan, Taimoor Muzaffar Gondal, Sheheryar Khan, Jia Wu, Muhammad Usman Hadi, John Heymach, Xiuning Le, Hong Yan, Tanvir Alam ▌论文网址: https://www.cell.com/heliyon/fulltext/S2405-8440(23)04783-7 ▌DOI: https://doi.org/10.1016/j.heliyon.2023.e17575 1974年,我们出版了首本旗舰期刊《细胞》。如今,CellPress已发展为拥有50多本期刊的全科学领域国际前沿学术出版社。我们坚信,科学的力量将永远造福人类。 CellPress细胞出版社 阅读原文 |
【本文地址】
今日新闻 |
推荐新闻 |