Science |
您所在的位置:网站首页 › 文山权威发布 › Science |
Science
|
特别提醒:墨卓生物微生物高通量单细胞基因组学技术——Microbe-seq合作正在全球征集,扫描文末二维码或点击阅读原文
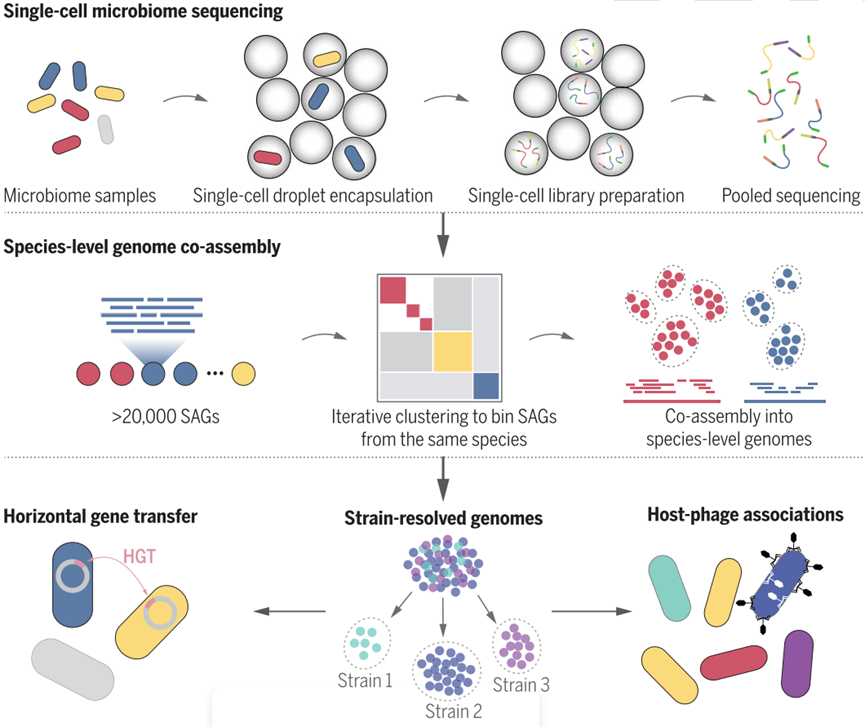
https://doi.org/10.1126/science.abm1483 近期,哈佛大学和麻省理工学院的研究团队在微生物群落研究方法学上取得重要突破,发明了微生物高通量单细胞基因组学技术——Microbe-seq。相关成果以研究长文(Research Article)的形式于6月3日在Science上以High-throughput, single-microbe genomics with strain resolution, applied to a human gut microbiome为题发表。 Microbe-seq技术集成了多种液滴微流控操作技术和定制开发的生物信息学分析手段,不需要培养即可从复杂微生物群落中获取成千上万个单细胞微生物的基因组信息,并组装出高质量的菌株水平基因组,从而能够在不损失分辨率或广泛物种适用性的基础上探究微生物群落的基因组。该方法应用面广泛,可用于具有复杂微生物群落的样本,如粪便、土壤和海洋等,在微生态研究中具有极大的市场应用潜力。 研究背景 微生物群落(microbiome)存在于各种不同的生态系统中,典型的微生物群落包括土壤、海洋或江湖等环境微生物以及人体或动物肠道微生物等。其中,人体肠道微生物(human gut microbiome)的组成和功能与人类的健康和疾病息息相关。每个个体的肠道微生物群组成都是特异的,虽然人们经常携带相似的微生物物种,但是不同个体有不同的菌株,不同菌株表现出巨大的基因组差异。菌株之间的这些基因组变异可能导致重要性状的差异,如抗生素耐药性、代谢能力以及与宿主免疫系统的相互作用,这可能对人类健康造成严重后果。 肠道微生物群落中的微生物行为不仅受特定菌株的存在影响,还与它们之间的相互作用密切相关,比如噬菌体调节细菌组成,以及微生物细胞之间遗传物质的转移。为了更好地理解这些微生物的影响,我们需要对特定微生物的基因和功能途径有详细的了解。 然而,要想阐明微生物菌株中的这些信息,可能会面临相当大的挑战。因为现有技术往往仅能在物种水平上对微生物进行分析,难以获知菌株水平上的重要差异。因此,如果能从微生物群落中解析出菌株水平的高质量基因组,那么这将极大地帮助我们理解对肠道微生物行为及其对健康的影响。 现有技术短板 01 目前宏基因组测序技术被广泛应用到探索复杂微生物群落,包括人类肠道微生物的组成,然而,宏基因组学通常无法解析菌株水平的基因组。 02 基于培养的方法只能偏向于容易培养的少数微生物菌株,且成本偏高。 03 基于微孔板的单细胞测序虽然可以产生菌株水平的基因组,但是成本等限制导致这种方法只能获得有限数量的微生物菌株。 技术和分析原理 为了解决上述问题,哈佛大学和麻省理工学院的研究团队开发了一种微生物高通量单细胞基因组测序技术Microbe-seq,这一技术的实验原理如下部分: 1、利用液滴微流控技术,研究团队将成千上万的微生物单独地包裹在液滴中; 2、在每个液滴中,裂解微生物并释放DNA; 3、进行全基因组扩增,将扩增出来的DNA打断并连上接头,随后在液滴中用带有特定序列的标签标记液滴内DNA; 4、将所有液滴内的DNA合并,进行建库测序。 由于液滴内DNA标记的标签对于每一个液滴都是不同的,因而将测序后的序列按照DNA标签进行区分,即可得到每一个液滴内单个微生物的基因组信息。这些具有相同barcode的测序序列的集合即称为一个single amplified genome (SAG)。 对于复杂微生物群落,其中大多数微生物的参考基因组一般是未知的。而单个SAG涵盖的基因组信息一般在全基因组的50%以内,无法直接被用于参考基因组。这为微生物组的单细胞基因组分析带来了一个独特的难题,为了得到高质量的微生物群落的参考基因组,研究团队还发展了一套通用的生信分析流程: 1、通过提取并比对各单细胞的基因组标志性信息(genomic signature),将样品中来自同一物种的单细胞微生物识别出来,然后合并在一起来组装出物种水平的参考基因组; 2、进一步将单个微生物的基因组和参考基因组对比,识别来自于不同菌株的单细胞微生物并进行基因组组装。 通过这种方法,研究人员获得了样品中多种不同物种及物种中不同菌株的基因组。
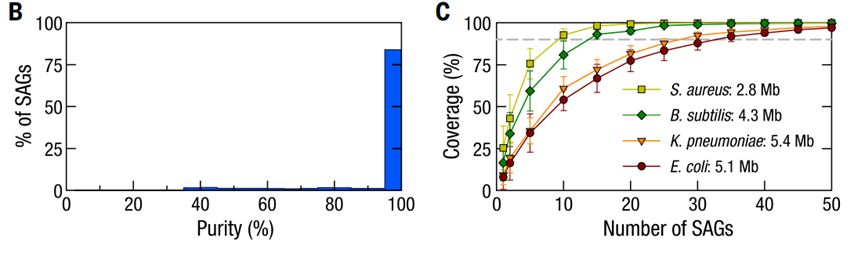
Microbe-seq概览 Microbe-seq性能评估 ——基于模拟微生物群落样本 为了表征每一个SAG所包含的基因组信息,研究人员用四种已知菌株构建了模拟微生物群落样本并应用Microbe-seq方法对其进行测试。这四种菌株包含了两种实验室常见的革兰氏阴性菌(大肠杆菌、肺炎克雷伯菌)和两种革兰氏阳性菌(枯草芽孢杆菌、金黄色葡萄球菌),经过实验,共得到了5497个SAG。为了验证单个SAG中的序列是否来自于单个微生物,研究人员用纯度来表征每个SAG中单微生物基因组的比例,SAG纯度的定义是一个SAG的所有能够比对到四种已知菌株的测序序列中,比对到四种菌株中的序列比例的最大值(即比对到上述四种菌株任一基因组的序列数/SAG比对到所有四种菌株的基因组总序列数的比值的最高值)。比对结果显示,在模拟样品中,84%(4612)的SAG纯度超过95%,表明绝大多数SAG都来自单一微生物(见下图B)。 研究人员同时也统计了单个SAG包含的基因组信息的比例,对于革兰氏阴性菌大肠杆菌和肺炎克雷伯菌,平均基因组覆盖率分别为8%和10%;而对于革兰氏阳性菌枯草芽孢杆菌和金黄色葡萄球菌,比例为17%和25%。为了获取完整的基因组信息,一种常用方法是合并来自相同物种的多个SAG的基因组信息。测试发现,对于上述四种菌株,仅需要几十个SAG就能覆盖基本完整的基因组(见下图C)。
基于模拟群落样本评估Microbe-seq性能 Microbe-seq应用 ——基于人体肠道群落样本 1、物种水平的高质量基因组组装: 研究人员将Microbe-seq方法应用于一个健康人捐赠的7份粪便样本,对应样品的宏基因组测序结果和分离培养鉴定结果在之前已经被报道,这些已经报道的工作为多维度评估Microbe-seq的结果提供了可能。通过Microbe-seq,研究人员从每个样本中得到了1000-7000个SAG,总数为21914个,每个SAG平均包含约70000条序列。 为了更好地对这些SAG进行组装,研究人员开发了一种不参考外部基因组的方法,将同一物种多个相似SAG进行共组装,然后将平均核苷酸同一性(ANI)值超过95%的基因组归为同一物种,并将它们的序列进行合并,进而获得物种水平的基因组。 组装获得的微生物基因组质量,一般用完整度和污染度两个指标进行评估:完整度>95%且污染度50%且污染度1000,引用8400余次(Google Scholar数据);申请国内专利5项(授权1项),PCT专利1项。承担并完成国家自然科学基金基金委重大研究计划培育项目、基金委新型冠状病毒应急攻关专项,担任国家重点研发计划课题负责人,中国科学院重点部署项目课题负责人;2020年3-4月代表微生物研究所在武汉参加抗疫攻关。 点评: 实现单个细胞精度的基因组学、转录组学,甚至甲基化组学、蛋白质组学和代谢组学的信息,有助于了解不同器官组织中细胞的组成、异质性和疾病中发生的改变;在真核生物尤其是人、各种模式动物中单细胞的多组学技术日趋成熟,并且一系列大的Single-Cell Atlas不断提供更完整的细胞多样性、新的疾病marker和潜在的治疗的靶点。然而微生物的单细胞组学相对来讲研究较少,其中有部分的技术因素例如通量问题,以及要解决的科学问题与真核生物往往不通。尤其是面向与人类健康、代谢和免疫息息相关的人体微生物组,绝大部分的研究工作还是使用bulk宏基因组测序的方法。Science此次发表的哈佛、麻省理工合作的新方法,极大的提高了单细胞基因组测序的通量,一次性获得成千上万个细胞的序列数据,并且拼接出了近百个物种的高质量基因组数据,以及物种内高度多样的菌株变异;在此基础上作者又深入的分析了菌株水平的水平基因转移事件(HGT),以及在单细胞水平获得了代表性噬菌体和宿主的关联信息。微生物群落的分析方法达到单细胞的精度,必然会对未来的微生物组的功能研究提供一个新的窗口,并且在物种、菌株、单细胞层面获得新的功能认知。这个方法未来有望继续结合空间信息、宿主提供的生态位,以及转录组甚至蛋白质组信息,继续深化对复杂微生物组的作用的理解。
戴磊 中科院深圳先进技术研究院 戴磊,中国科学院深圳先进技术研究院研究员、博士生导师,合成微生物组研究中心主任。国家重点研发计划青年项目负责人,入选《麻省理工科技评论》中国区“35岁以下科技创新35人”。戴磊实验室在定量生物学、合成生物学与微生物组学的交叉领域进行原创性研究,致力于对宿主共生微生物组的结构和功能进行精准表征和调控,解决人体健康、农业生产等重大问题。研究成果以通讯作者或第一作者发表在Science、Nature、PNAS、ISME Journal、ACS Synthetic Biology等国际学术期刊。 点评: 微生物组学领域的科学突破离不开技术创新。David Weitz课题组与Eric Alm课题组强强联合,整合液滴微流控技术和生物信息学分析,开创了适用于复杂微生物群落的单细胞基因组测序分析方法Microbe-seq。这是一项非常漂亮的工作,具有很高的科学意义和应用价值。Microbe-seq技术可以高通量获取单个样本中几千甚至更多个微生物细胞的高质量基因组,进而深入研究菌株水平上的功能差异。以人体肠道微生物组为例,该研究展示了菌株水平的基因组分析对于研究微生物生态和进化的重要性,在水平基因转移、噬菌体互作等方面带来新的认知。在不久的将来,单细胞技术必将成为微生物组研究不可或缺的利器,助力复杂微生物群落的精准解析与改造。
晁彦杰 中国科学院上海巴斯德研究所 晁彦杰,中国科学院上海巴斯德研究所研究员、课题组长,入选国家高层次海外人才计划。博士毕业于德国洪堡大学和马普感染生物学研究所,在德国维尔茨堡大学和美国塔夫茨大学完成博士后研究。主要从事微生物RNA测序技术和原核非编码RNA的功能研究,曾与2020年诺贝尔化学奖得主Emmanuelle Charpentier合作在细菌中首次发现了CRISPR-Cas9系统,为基因组编辑提供了核心工具。近年来在Nature, Molecular Cell, EMBO Journal, EMBO Molecular Medicine等国际权威期刊发表论文20多篇,多次受到杂志社撰文点评、F1000推荐或被评为高被引论文,被引用5000余次。兼任BMC Genomics杂志编委和mBio等20多家权威SCI期刊的评审专家。课题组欢迎有志于微生物学研究的博士后、研究生和联培生前来进修学习和工作,干湿技术皆可,长期有效。 点评: 这是一次技术上和维度上的突破,在单细胞水平实现了对人体微生物群落的基因组测序、组装与分析。虽然真核细胞研究已经全面迈进了单细胞组学时代,但是原核微生物的单细胞分析仍处于萌芽初期。这主要是因为微生物的细胞体积和基因组比动物细胞小几个数量级,微生物细胞的分离、裂解,核酸的捕获与扩增等环节都存在较大的技术难度。这篇文章利用微流控技术,实现了微生物单细胞的分离与分析,将每个细菌包裹在单个液滴中进行裂解和建库,实现了高通量的单细菌基因组测序。 为展现这个技术的能力与先进性,作者首次将Microbe-seq应用于人体肠道微生物群这样一个复杂的微生态系统,把肠道微生物分析推进到了菌株水平。通过对一个人体的多份粪便样品进行系统分析,作者鉴定出了上万个细菌基因组(SAG),覆盖了20多个种属。对于没有参考基因组的细菌,利用从头组装获得了几十个高质量的接近于完整的基因组序列,实现了不依赖于细菌纯培养的基因组测序。 值得注意的是,虽然在本工作中,作者在一定程度上展示了Microbe-seq方法对不同类型细菌的捕获的均匀性。例如在四个菌株的模拟样品测试中捕获到数量接近的单细胞数量。然而,Microbe-seq反应的变形菌门和拟杆菌门细菌的丰度相对宏基因组偏低,有必要用一个已知组分和含量的复杂模拟样品对该方法进行系统验证测试。此外,该方法对革兰氏阴性菌的基因组覆盖度相对于革兰氏阳性菌偏低,可能与选用的裂解方法有关,通过对裂解技术的进一步优化,另外利用二代与三代测序技术的结合,相信该技术将能够产生更多更完整的基因组序列。 展望未来,Microbe-seq技术将有极大的科研潜力和应用价值。单细胞基因组分析未来有望扩展到更大的人群和样品队列、覆盖多种不同的疾病、增加时间和空间的维度,也有希望实现对肠道细菌、真菌和病毒等物种进行同时分析,有助于更准确地发现关键微生物或者疾病标志物。未来通过技术升级,增加对液滴或细菌的筛选和分选,该技术也将有助于分离特定的菌株进行培养组学研究,或者对特定微生物群体进行更有针对性的研究与鉴定。
扫描二维码或点击阅读原文,申请Microbe-seq合作 猜你喜欢iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAPiMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKlaiMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测 10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature 系列教程:微生物组入门 Biostar 微生物组 宏基因组 专业技能:学术图表 高分文章 生信宝典 不可或缺的人 一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote 扩增子分析:图表解读 分析流程 统计绘图 16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun 生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘 写在后面为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组” 点击阅读原文 |





【本文地址】