中国医药研发创新专题报告【30页】 中国医药企业研发投入持续增加医药行业研发费用持续上升制药工业上市公司的研发投入持续上升,2021年国内A股上市制药工业企... |
您所在的位置:网站首页 › 我国上市药企 › 中国医药研发创新专题报告【30页】 中国医药企业研发投入持续增加医药行业研发费用持续上升制药工业上市公司的研发投入持续上升,2021年国内A股上市制药工业企... |
中国医药研发创新专题报告【30页】 中国医药企业研发投入持续增加医药行业研发费用持续上升制药工业上市公司的研发投入持续上升,2021年国内A股上市制药工业企...
|
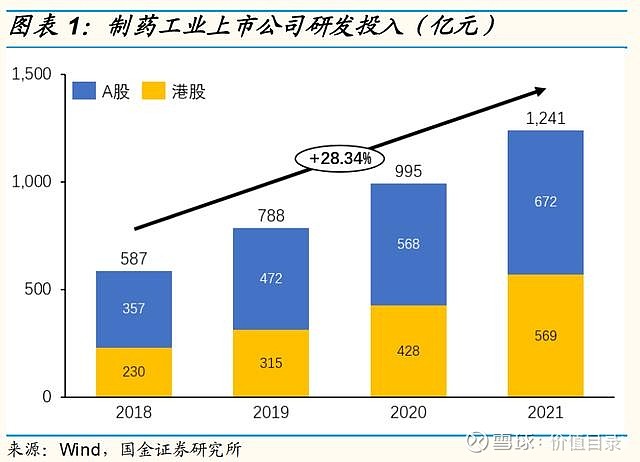
来源:雪球App,作者: 价值目录,(https://xueqiu.com/7697110006/221344309) 中国医药企业研发投入持续增加医药行业研发费用持续上升 制药工业上市公司的研发投入持续上升,2021年国内A股上市制药工业企业的研发费用达到1241亿元,其中A股上市企业研发投入达到672亿元,港股上市企业研发投入达到569亿元。2018奶奶以来研发费用的复合年增长率达到28.34%。制药工业上市企业研发费用率也快速上升。2018年A股和港股的制药工业企业的研发投入分别为4.6%和6.1%,到2021年分别增长到了7.0%和10.9%。但是,与美股上市的制药企业相比,国内企业的研发投入还有很大的增长空间。
分版块来看,A股化药公司(SW-化学制剂)的研发费用总额从2018年的171亿元持续增长到2021年的317亿元,研发费用率从2018年的4.93%上升到2021年的7.81%。A股中药公司(SW-中药II)的研发费用总额从2018年的60亿元持续增长到2021年的87亿元,研发费用率从2018年的2.05%上升到2021年的2.63%。A股生物药公司(SW-其他生物制品)的研发费用总额从2018年的81亿元持续增长到2021年的175亿元,研发费用率从2018年的19.77%上升到2020年的40.54%,2021年研发费用率下降至32.63%。2020年至2021年的费用率波动可能与2020年总营收下降10%,而2021年总营收增长42%有关,整体研发费用率呈增长趋势。 港股化药公司(SW-化学制剂)的研发费用总额从2018年的91亿元持续增长到2021年的188亿元,研发费用率从2018年的9.58%上升到2021年的14.40%。港股中药公司(SW-中药II)的研发费用总额从2018年的12亿元持续增长到2021年的17亿元,研发费用率从2018年的1.83%下降到2021年的1.68%,是唯一研发费用率下降的医药板块。港股生物药公司(SW-其他生物制品)的研发费用总额从2018年的26亿元持续增长到2021年的91亿元,研发费用率从2018年的20.84%上升到2020年的31.51%,2021年研发费用率下降至28.95%。2020年至2021年的费用率波动可能与2021年总营收增长40%有关,整体研发费用率呈增长趋势。 与美股医药企业的研发费用率相比,A股和港股生物药企的研发费用率均还有增长空间,港股化药企业与美股化药企业的研发费用率已经十分接近,A股化药企业的研发费用率仍然有提升空间。
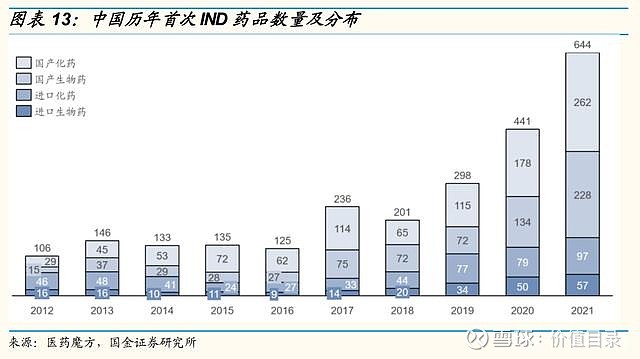
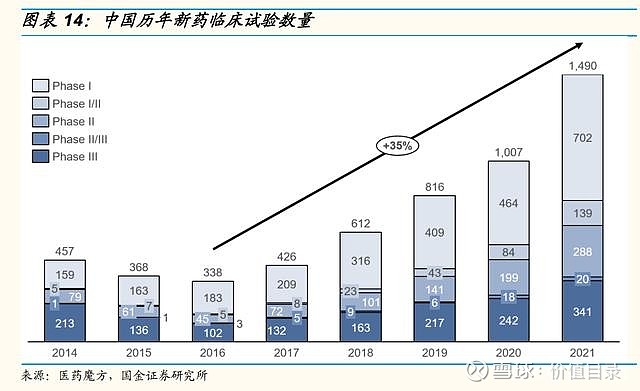
研发支出资本化方面,A股制药工业整体保持稳中有降的趋势。分版块来看,化药、生物药、重点创新药企业的趋势基本与整体行业一致,化药板块的研发支出资本化率较高。 重点创新药企业分市值来看,市值千亿以上的四家公司整体资本化率较低,百济神州和君实生物的研发支出未进行资本化处理,恒瑞医药从2021年起进行3期及关键性注册临床的研发支出资本化处理,2021年的资本化率为4%,复星医药历年资本化率较高,但也从2018年的41%下降到了2021年的23%。市值百亿至千亿的中型创新药公司的研发支出资本化率差异较大,传统型企业如亿帆医药、信立泰、海思科研发支出资本化率较高,而biotech企业如神州细胞、荣昌生物、上海谊众未进行资本化处理。市值百亿以内的创新药企业大部分未进行资本化处理。 研发人员数量及薪酬持续增加 A股上市公司的研发人员总数和研发薪酬支出均呈增长趋势。为了踢除部分公司披露不完整的影响,我们选取了连续四年有相关数据披露的公司(生物药公司和重点创新药公司的研发人员选取了连续三年有数据披露的公司),统计了调整后的研发人员和研发薪酬支出,调整后数据显示,除中药企业的研发人员在2021年有小幅度回调以外,其它几类公司的研发人员数量,以及四类公司的研发薪酬支出均呈持续增长。2021年研发薪酬支出的增长尤为明显,各类公司的研发人员平均薪酬也呈稳定增长趋势。 IND数目,临床试验数量 在持续的高研发投入下,中国国产药物的IND数量迅速上市,国内开展的临床试验数量也大幅增加。2018年之后,中国首次IND新药数量快速上升,近3年总体CAGR为47%,其中,国产创新药首次IND新药数量增长显著高于进口创新药,2021年国产药品占比达76%,化药和生物药均保持高速增长。2021年,新兴疗法如PROTAC、三抗/四抗、CAR-NK、肿瘤浸润淋巴细胞(TIL)等首次在中国IND。 2017年之后,临床登记数量也迅速增长,2021年临床登记数量达1490个,同比增长接近50%。从2016年到2021年的临床试验数量的年复合增长率达到35%。
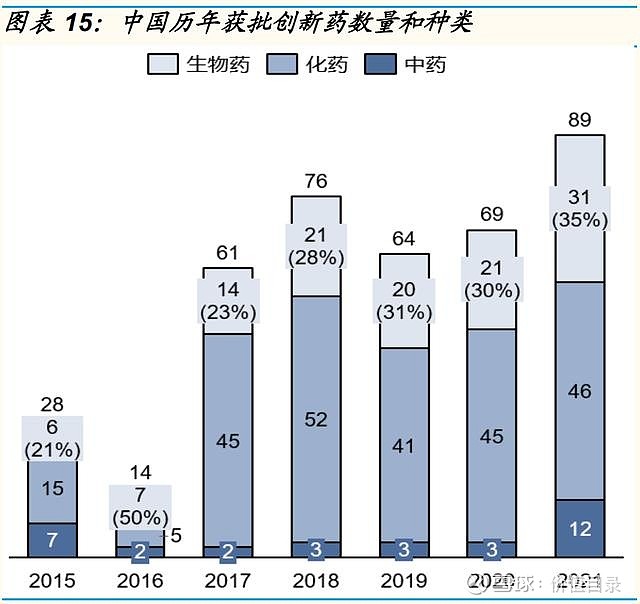
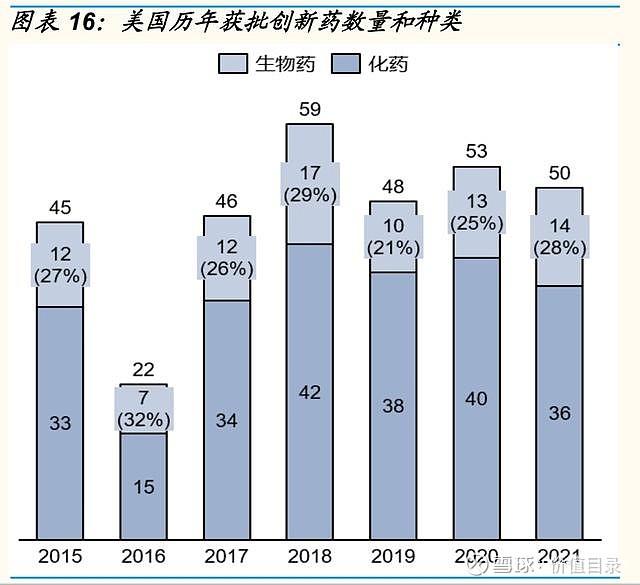
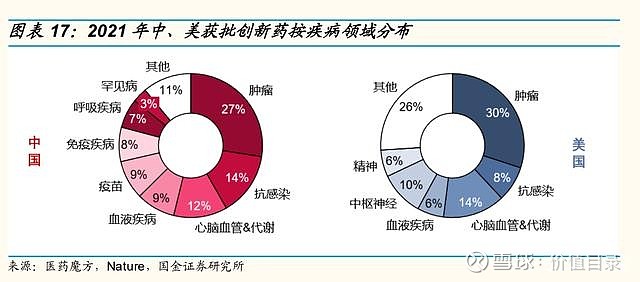
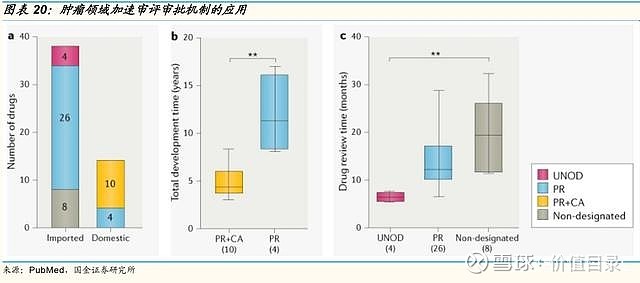
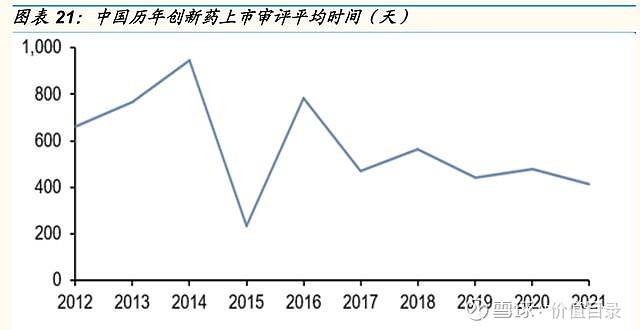
2021年中国获批上市的创新药数量和种类与美国相当 2017年以来,国内历年获批的创新药数量呈增长趋势,2021年国内共有89款创新药获批,其中生物药创新药31款,化药创新药46款,中药创新药12款。从绝对值上看,中国每年获批的创新药数量已经与美国相当。从生物药和化药的分布来看,也与美国的分布类似。从获批创新药的治疗领域来看,中国和美国也具有相似性,2021年中、美获批创新药中占比最多的均为肿瘤领域,分别占比27%和30%。心血管代谢、抗感染、血液疾病也是中、美新上市药物较为集中的领域。不同之处在于,2021年美国有10%的创新药为中枢神经领域,而中国获批的中枢神经领域创新药数量较少。 中国创新药加速审批路径日益成熟 美国目前拥有4中加速审评审批的路径,包括优先审评(PriorityReview,PR)、加速审批(AccelerateApproval)、快速通道(FastTrack)和突破性疗法(BreakthroughTherapyDesignation)。 中国近年的药政改革也引入了一系列药品加快审评审批路径,以加速药物开发和审评。这些措施中,包括2015年实施的对于具有显著临床获益药物缩短审评时间的优先审评(PriorityReview)制度,2017年国家药品监督管理局发布实施的允许以替代终点或中间临床终点指标支持用于治疗严重或危及生命疾病的药物获得附条件批准(ConditionalApproval)的相关法规,以及NMPA在2018年发布的允许基于其他国家/地区试验数据加速批准境外上市的临床急需新药在我国上市。2020年7月,我国继续推出一项突破性疗法(BT)路径,授予初步证据显示具有明显临床优势的产品。获得认定的药物可在开发和注册中得到监管机构的深入指导。 加速审评审批路径的不断丰富,加快了创新药在中国的上市时间。以肿瘤领域为例,2016-2020年,国内共批准了52种肿瘤创新药,其中38个为进口肿瘤创新药,14个为国产肿瘤创新药。14个国产肿瘤创新药中,10个通过优先审批程序和附条件批准上市,其进入临床开发到首次获批上市的中位时间为4.3年,而另外4个肿瘤创新药仅有优先审评、没有附条件批准,其进入临床开发到首次获批上市的中位时间为11.3年。38个进口肿瘤创新药中,4个通过临床急需境外新药路径在国内上市,中位审评审批时间为6.4个月,26个通过优先审评路径在国内上市,中位审评审批时间12.2个月,而8个未通过加速审评审批路径的进口创新药的中位审评审批时间长达19.4个月)。 在日益完善的审评审批体系和逐渐丰富的加速审评审批路径下,中国创新药的平均上市审评时间呈逐渐缩短趋势。据医药魔方统计,2021年获批创新药平均审评时间约为412天。
创新药中美获批时间差缩短 受这些政策变化的激励,2010-2020年间中国批准的43种进口肿瘤新药相比美国上市时间的差距显著缩短,2006-2010年在美国批准的进口肿瘤药的中美上市滞后时间中位数为8.7年,而2016-2020年在美国批准的肿瘤药物的滞后时间仅为2.7年。 我们选取了肿瘤领域的4个代表性的分子类型及靶点的药物,比较了firstin-class新药在美国上市的时间、在中国上市的时间、以及国内首款同靶点国产药上市的时间。 从小分子的代表性靶点EGFR来看,吉非替尼在美国上市时间为2003年2月,在国内上市时间为2004年12月,时间相差22个月,而首个国产EGFR为贝达药业的埃克替尼,在2011年6月获批上市,距离吉非替尼在国内上市已经过去77个月。 从单抗来代表药物PD-1来看,帕博利珠单抗在美国的上市时间为2014年9月,在中国上市时间为2018年7月,时间相差46个月,而此后首个国产PD-1,君实生物的特瑞普利单抗在2018年12月获批上市,距离帕博利珠单抗在国内上市仅过去了6个月。 从ADC代表药物HER2ADC来看,恩美曲妥珠单抗在美国上市时间为2013年2月,在国内上市时间为2020年1月,时间相差83个月。而首个国产ADC,荣昌生物的维迪西妥单抗在国内上市时间为2021年6月,距离恩美曲妥珠单抗在国内上市仅过去了17个月。 从细胞疗法代表药物CAR-T来看,阿基仑赛在美国上市时间为2017年10月,在国内上市时间为2021年月6,时间相差44个月。而首个国产CAR-T疗法,药明巨诺的瑞基奥仑赛在2021年9月就获批上市,距离阿基仑赛国内上市仅过去3个月。
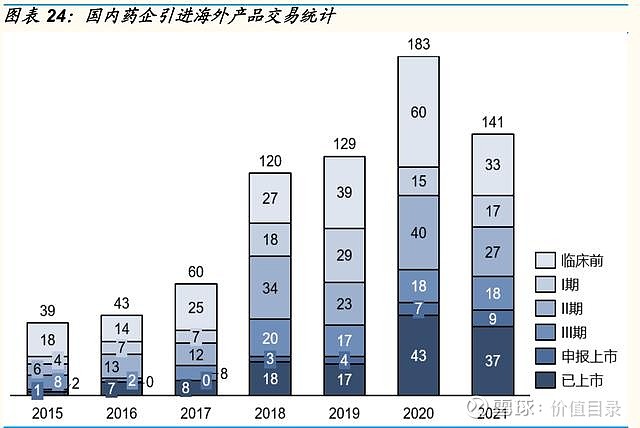
引进海外创新药 产品引进可以帮助药企快速补充管线,实现弯道超车,是大型跨国药企常用的交易方式。总体来看,近年来,中国创新药企引进海外产品的数量呈持续上升趋势,尤其对于临床前的早期产品及已上市的成熟产品引进数量大幅增加。引进海外产品的交易数量在2020年达到183项的高峰。 2021年国内药企引进海外创新药的交易数量回调,但其中,不少国内药企在一年之内引进了3项或以上产品,也有不少金额较大的大型交易。再鼎医药在2021年引进了5款产品的权益,并持续推进研发进展。 2021年1月6日,再鼎医药从argenx引进了自身免疫药物efgartigimod在大中华区的开发和商业化权益,再鼎医药将负责efgartigimod多个适应症在中国的全球注册临床研究开发工作,也将在大中华区启动多个新适应症的2期验证性研究,以在全球范围内加速开发efgartigimod的更多自身免疫类适应症。再鼎医药将支付1.75亿美元的合作付款,其中包括7500万美元的预付款,以再鼎医药增发股票形式支付,7500万美元临床开发成本共担付款,以及潜在的efgartigimod在美国的注册里程碑付款和大中华区的年净销售额分成。 2021年1月11日,再鼎医药扩大与TurningPoint的合作,引进MET/SRC/CSF1R抑制剂TPX-0022在大中华区独家开发和商业化权利,再鼎医药将支付2500万美元的首付款,以及潜在的不超过3.36亿美元的开发、注册和销售的里程碑付款和TPX-0022在大中华区的销售提成。2021年11月,再鼎获得了TPX-0022在国内的IND批件,用于治疗MET基因变异的局部晚期或转移性非小细胞肺癌、胃癌或实体瘤。此前2020奶奶7月,再鼎医药已经从TurningPoint引进了repotrectinib在大中华区的独家权益。 2021年6月1日,再鼎医药从Mirati引进了KRASG12C抑制剂adagrasib在大中华区的开发和商业化的独家权益,再鼎医药将支付6500万美元的首付款,以及潜在的不超过2.73亿美元的开发、注册和销售的里程碑付款和TPX-0022adagrasib在大中华区的销售提成。2022年1月19日,再鼎医药获得了adagrasib在国内的IND批件。 2021年11月9日,再鼎医药从Blueprint引进了新一代EGFR抑制剂BLU-945和BLUE-701在大中华区的开发和商业化的独家权益。再鼎医药将支付2500万美元的首付款,及潜在里程碑付款和销售提成。 同样在2021年11月9日,再鼎医药从Karuna引进了口服M1/M4型毒蕈碱激动剂KarXT在大中华区的开发、生产、和商业化的独家权益。再鼎医药将支付3500万美元的首付款,以及潜在的不超过1.52亿美元的里程碑付款和百分之十几至百分之二十之间的销售提成。
2021年联拓生物引进了5款产品: 2021年3月2日,联拓生物从ReViral引进治疗呼吸合胞病毒(RSV)药物sisunatovir在中国大陆、香港、澳门和新加坡地区共同开发和商业化的独家权益。联拓生物将支付1400万美元的首付款,以及潜在的不超过1.05亿美元的开发和商业里程碑付款和许可地区两位数的销售提成。合作伙伴ReViral致力于RSV的抗病毒疗法,sisunatovir是其主要的在研产品,2022年4月7日,辉瑞宣布收购ReViral公司,包括前期和开发里程碑付款在内的收购金额可高达5.25亿美元。 2021年3月29日,联拓生物从Tarsus引进眼科疾病药物TP-03(洛替拉纳滴眼液)在大中国区的开发和商业化权益。联拓生物将支付1500万美元的首付款,以及潜在的不超过1.85亿美元的开发和商业化里程碑付款和许可区域的销售提成。 2021年5月11日,联拓生物从Nanobiotix引进放射增强剂NBTXR3(二氧化铪纳米颗粒)在大中华区、韩国、新加坡和泰国的开发和商业化的权益。联拓药业将支付2000万美元首付款,以及潜在的不超过2.2亿美元的开发和商业化里程碑付款和许可区域的销售提成。2022年4月20日,联拓生物获得NBTXR3在国内的IND默示许可,拟开发用于治疗头颈部鳞状细胞癌。 2021年5月17日,联拓生物从Landos引进LANCL2通路激活剂Omilancor(BT-11)和NLRX1靶点的NX-13两款药物在大中华区和亚洲特定市场的开发和商业化独家权益。联拓生物将支付1800万美元首付款,以及潜在的不超过2亿美元的开发和商业化里程碑付款和许可区域的销售提成。 2021年6月2日,联拓生物从Lyra引进鼻内抗炎药物LYR-210在大中华区、韩国、新加坡和泰国的开发和商业化的独家权益。联拓生物 将支付1200万美元首付款,以及潜在的不超过1.35亿美元的开发和商业化里程碑付款,和许可区域的销售提成。
2021年先声药业引进了3款产品: 2021年3月29日,先声药业从Kazia引进PI3K/mTOR抑制剂pazalisib在大中华区的开发和商业化独家权益。先声药业将支付首付款、里程碑付款和许可区域的销售提成,具体金额未披露。2021年6月29日,先声药业从Vivoryon引进靶向神经毒性淀粉样蛋白N3pE(pGlu-Aβ)的口服小分子谷氨酰肽环转移酶(QPCT)抑制剂Varoglutamstat(PQ912,SIM0408),和处于临床前研究阶段的单克隆N3pE抗体PBD-C06。2022年2月28日,Varoglutamstat在中国获得临床试验批准。2021年12月15日,先声药业从Avilex引进靶向突触后密度蛋白95(PSD-95)的脑卒中创新药AVLX-144在大中华地区的开发、生产和商业化的权益。先声药业将支付首付款,及潜在的超过1.75亿美元的开发和商业化里程碑付款,和许可区域内的销售提成。 2021年齐鲁药业引进了3款产品: 2021年2月16日,齐鲁制药从CendTherapeutics引进靶向α-v的环肽CEND-1在大中华区的独家权益。齐鲁药业将支付1000万美元首付款,以及不超过2.25亿美元的里程碑付款,和许可区域的销售提成。2021年7月,CEND-1联合吉西他滨和白蛋白紫杉醇用于一线治疗晚期转移性胰腺癌的临床试验申请获得CDE的默示许可。2021年4月16日,齐鲁制药从Peptron引进靶向MUC1的抗体偶联药物PAb001-ADC在全球开发、生产、销售和商业化的独家权益。2021年12月13日,齐鲁制药从Arbutus引进了RNA干扰药物AB729在大中华区的开发和商业化的独家权益。齐鲁将向Arbutus支付4000万美元首付款和1500万美元的股权投资,以及潜在的不超过2.45亿美元的里程碑付款,和许可区域的销售提成。5月10日,AB729用于治疗或预防乙型肝炎(HBV)的临床试验申请获得CDE受理。 2021年9月13日,云顶新耀从Providence引进了新冠mRNA疫苗PTXCOVID19-B在中国及部分亚洲新兴市场的权益,云顶新耀将支付5000万美元的首付款,以及潜在的销售提成。同时,云顶新耀还与Providence建立战略合作关系,利用其mRNA技术平台在全球开发更多mRNA产品。云顶新耀将支付5000万美元首付款,以及潜在的不超过3亿美元的里程碑付款。 我们认为,近年来从海外市场引起创新药产品在国内的开发和商业化权益已经愈发成为药企补充管线的重要来源,这也进一步加速了海外研发阶段领先的新型创新药在国内的研发进展和可及性,有利于中国加速中国创新研发与国际领先水平的进一步接轨。
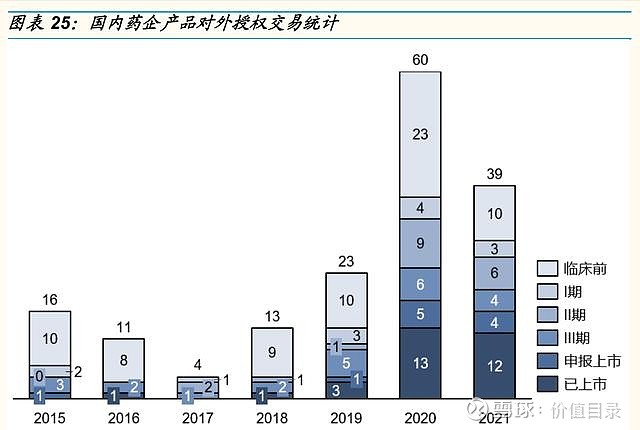
自研创新药对外许可 中国药企通过对外许可,将自主研发的产品的海外权益许可给大型跨国药企或海外本土药企,则可以借助合作伙伴在海外的研发、申报、销售经验,加速实现产品在海外的上市,补充企业现金流。 2021年1月12日,百济神州与诺华达成PD-1产品替雷利珠单抗的合作与授权协议,诺华获得替雷利珠单抗在美国、加拿大、墨西哥、欧盟成员国、英国、挪威、瑞士、冰岛、列支敦士登、俄罗斯和日本等多个国家的开发、生产和商业化权利。双方将在上述国家对替雷利珠单抗进行共同开发,其中诺华将在过渡期后负责注册申请,并在获得批准后开展商业化活动。此外,双方均可在全球范围内开展临床试验以评估替雷利珠单抗联合其他抗肿瘤疗法的潜在用药组合;百济神州可在北美地区共同进行产品销售,其中部分运营资金将由诺华提供。根据协议,百济神州将获得6.5亿美元的首付款,以及潜在的13亿美元的注册里程碑付款和2.5亿美元的销售里程碑付款,和替雷利珠单抗在授权地区未来销售的销售提成。 2021年12月21日,百济神州扩大了与诺华的合作,进一步将其TIGIT抑制剂ociperlimab纳入合作,向诺华授予独家的、基于时间的选择权,诺华行使该选择权可以获得在美国、加拿大、墨西哥、欧盟成员国、英国、挪威、瑞士、冰岛、列支敦士登、俄罗斯和日本对ociperlimab进行开发、生产和商业化的独家许可。根据协议,百济将获得3亿美元首付款,如果诺华行使选择权,则百济有资格获得6亿美元或7亿美元的额外付款,和潜在的7.45亿美元注册里程碑付款和11.5亿美元销售里程碑付款,以及许可地区20%-25%的销售提成。 2021年2月10日,君实生物与Coherus达成PD-1产品特瑞普利单抗的授权许可协议,授予Coherus在美国和加拿大开发和商业化特瑞普利单抗的独家权益,以及TIGIT单抗JS006和IL-2药物JS018-1的选择权、2个早期检查点抑制剂抗体药物的优先谈判权。根据协议,君实生物获得1.5亿美元的首付款。对于特瑞普利单抗项目,在达到相应的里程碑事件后,君实生物将获得累计不超过3.8亿美元的里程碑款,外加许可区域内特瑞普利单抗年销售净额20%的销售分成。 2022年1月10日,君实生物与Coherus扩大合作范围,Coherus已启动行使TIGIT单抗JS006在美国和加拿大的许可选择权的程序。Coherus将就JS006的合作向君实生物支付3500万美元首付款,最高达2.55亿美元的开发、申报和销售里程碑付款,以及产品销售净额18%的销售分成。 2021年8月8日,荣昌生物与Seagen达成HER2ADC产品维迪西妥单抗的授权许可协议,荣昌生物保留Seagen在大中华区、亚洲除日本新加坡以外地区的权益,授予Seagen全球其他地区的独家开发及商业化维迪西妥单抗的权益。荣昌生物获得首付款2亿美元,以及潜在的不超过24亿美元的里程碑付款,和产品在许可地区的高个位数至百分之十几的销售提成。 2021年7月13日,诺诚健华与Biogen达成BTK抑制剂奥布替尼的授权合作协议,诺诚健华授予Bioegen奥布替尼在多发性硬化症的全球权利,及除中国(含香港、澳门、中国台湾)以外区域的某些自身免疫性疾病领域独家权益。诺诚建华获得1.25亿美元首付款,以及潜在的8.125亿美元的里程碑付款和百分之十几销售分层。 中国创新药企对外授权在近几年迅速增加,我们认为,2021年以来几项大金额的授权交易,尤其是百济神州与诺华、君实生物与Coherus的二度合作,反映出中国药企的研发实力进一步受到海外认可。
国产创新药在美国申报上市情况梳理 自2019年11月百济神州的BTK抑制剂泽布替尼在美国获批上市以来,不断有国产创新药向美国FDA提交上市申请,但结果不尽相同。除泽布替尼以外,传奇生物的CAR-T疗法cilta-cel获得上市批准,而万春医药的普那布林、信达生物的信迪利单抗、和黄医药的索凡替尼均需要再补充临床试验,君实生物的特瑞普利单抗则需要变更质控流程。总体来看,国产创新药在美国申报上市仍处于规则探索阶段。 百济神州泽布替尼:2019年11月15日,百济神州宣布泽布替尼获得美国FDA上市批准,用于治疗既往接受过至少一项疗法的成年套细胞淋巴瘤(MCL)患者。FDA此项批准是基于两项临床试验的有效性数据,数据显示泽布替尼在参与两项临床试验的患者中均产生高达84%的ORR。在泽布替尼用于治疗复发/难治性(R/R)MCL患者的多中心2期临床试验BGB-3111-206中,ORR为84%,包括59%的完全缓解以及24%的部分缓解。此项试验的中位持续缓解时间为19.5个月,中位随访时间为18.4个月。在全球1/2期临床试验BGB-3111-AU-003中,ORR为84%,包括22%的完全缓解以及62%的部分缓解。此项试验的中位DOR为18.5个月,中位随访时间为18.8个月。 万春医药普那布林:2021年12月1日,万春医药宣布FDA对普那布林联合粒细胞集落刺激因子(G-CSF)用于预防化疗引起的中性粒细胞减少症(CIN)的新药申请(NDA)发出了完全回复函(CRL)。FDA指出,单一注册试验的结果不足以证明效益,FDA建议进行第二项对照试验以满足支持CIN适应症的实质性证据要求。此前的2021年4月,万春医药基于普那布林的国际多中心III期临床试验PROTECTIVE-2研究结果,同步向中美提交了上市申请。PROTECTIVE-2国际多中心III期临床研究是一项在接受TAC(多西他赛、阿霉素和环磷酰胺)化疗的乳腺癌患者中比较普那布林(40mg)联合培非格司亭(6mg)治疗和培非格司亭单药疗效的III期全球多中心、双盲、阳性对照临床研究。研究结果显示,联合治疗组和培非格司亭单药组未发生4级中性粒细胞减少症的患者百分比分别为31.5%和13.6%,具有显著性差异。
信达生物信迪利单抗:2022年2月10日,FDA肿瘤药物咨询委员会(OncologicDrugsAdvisoryCommittee,ODAC)开展会议讨论礼来和信达的PD-1药物信迪利单抗治疗非小细胞肺癌的上市申请。此项申请是基于信迪利单抗治疗非小细胞肺癌的中国3期临床ORIENT-11试验,最终独立调查小组以14:1的投票结果反对此项BLA申请,并宣布在批准之前,必须进行另一项临床试验,证明该药对美国患者有效。2022年3月24日,礼来宣布收到美国FDA签发的关于信迪利单抗联合培美曲塞和铂类用于非鳞状非小细胞肺癌一线治疗的新药上市申请的完全回复信(CompleteResoinseLetter,CRL)。这也意味着FDA未批准该项上市申请。 传奇生物cilta-cel:2022年2月28日,传奇生物靶向B细胞成熟抗原(BCMA)的CAR-T疗法西达基奥仑赛(cilta-cel)的上市申请正式获FDA批准,用于治疗复发/难治性多发性骨髓瘤(r/rMM)患者。FDA此次批准是基于一项关键性Ib/II期CARTITUDE-1研究结果,该研究招募的97例受试者中,评估了cilta-cel在复发/难治性多发性骨髓瘤患者中的疗效和安全性,研究结果显示,中位随访12.4个月时,独立委员审查的ORR为97%,包括67%的sCR(严格的完全缓解),92.8%的患者获得了非常好的部分缓解(VGPR)及以上。21.7个月随访数据显示,83%的患者达到了sCR,95%的患者获得了VGPR及以上。PFS和OS尚未达到,2年PFS为61%,2年OS为74%。2022年3月24日,杨森/传奇生物宣布欧洲药品管理局(EMA)人用药品委员会(CHMP)对Carvykti(西达基奥仑赛,cilta-cel)的欧洲上市许可给予推荐,用于治疗既往接受过至少三种治疗,包括免疫调节药物、蛋白酶体抑制剂和抗CD38抗体,并且末次治疗出现疾病进展的复发或难治性多发性骨髓瘤成人患者。 君实生物特瑞普利单抗:2022年5月2日,君实生物宣布,美国FDA寄发了一封关于PD-1特瑞普利单抗联合吉西他滨╱顺铂作为晚期复发或转移性鼻咽癌患者的一线治疗和单药用于复发或转移性鼻咽癌含铂治疗后的二线及以上治疗的BLA申请的完整回复信。FDA要求进行一项质控流程变更。君实计划与FDA直接会面,并预计于2022年仲夏之前重新提交该BLA。同时,待完成的现场核查因新型冠状病毒肺炎疫情相关的旅行限制而受阻,具体现场核查时间将另行通知。此前2021年3月,君实生物正式向FDA滚动提交了特瑞普利单抗用于治疗复发或转移性鼻咽癌的BLA并获得FDA滚动审评,特瑞普利单抗成为首个向FDA提交BLA的国产抗PD-1单抗。上市申请申请是基于鼻咽癌一线适应症的JUPITER-02试验,和鼻咽癌二线及以上适应症的POLARIS-02试验,两项临床试验区域分别在亚太地区和中国开展,并未纳入美国患者。此外,在美国目前尚无免疫疗法获批用于治疗鼻咽癌。 和黄医药索凡替尼:2022年5月2日,和黄医药宣布,美国FDA已就索凡替尼用于治疗胰腺和非胰腺神经内分泌瘤的新药上市申请发出完整回复函。索凡替尼的安全性和疗效已在两项在中国晚期胰腺和非胰腺神经内分泌瘤患者中开展的随机双盲的III期研究(SANET-ep和SANET-p研究)中得到证明。一项在美国开展的桥接研究结果亦显示出与两项SANET研究人群中相似的安全性及疗效。FDA认为当前基于两项成功的中国III期研究以及一项美国桥接研究的数据包尚不足以支持药品现时于美国获批。该完整回复函中表明,需要纳入更多代表美国患者人群的国际多中心临床试验(MRCT)来支持美国获批。
信迪利单抗ODAC会议的启发 国产创新药出海仍处于规则探索阶段,但是信迪利单抗的ODAC会议上,FDA对获批要求的详尽讨论,给了后续创新药在美国申报上市的重要启发。 主张进行国际多中心临床试验(Multi-RegionalClinicalTrial,MRCT)。FDA指出,国际人用药品技术要求协调理事会(InternationalCouncilforHarmonisationofTechnicalRequirementsforPharmaceuticalsforHumanUse,ICH)对药品上市的有效性数据的要求,经过20年的发展,已经从过去的序贯性地在不同地区做桥接试验,发展为了主张进行国际多中心临床试验,并在国际多中心临床试验中考虑不同地区的差异。大部分美国的药品上市申请都是基于国际多中心临床试验,它便于评估药品在不同地区和不同人群之间的一致性,同时也有利于药物更快的在全球达到可及性。 特定情况下可以接受纯外国数据。根据ICH和美国监管规定的要求,在以下三种要求都满足的情况下,可以仅基于外国人的临床数据给予上市批准: (1)适用性:临床试验设计和数据适用于美国人群和美国临床实践;(2)研究由具有公认能力的临床研究者进行;(3)临床数据具有可靠性,FDA认为不需要核查或通过FDA现场核查或通过FDA其他方式的核查; 其中,适用性的内在要求较高。适用性要求当中,包含了适用美国人群和适用美国临床实践两部分。其中,适用于美国人群就包含了试验人群在基线方面需要与美国人群类似,包括人种及人种多样性、年龄分布、性别分布、体重、抽烟比例等多个方面。从这一点来看,仅在中国完成的注册临床试验很难达到适用美国人群的要求。其次,适用美国临床实践提出了更高的要求。一方面临床试验的终点设计要与美国在该领域批准的新药的终点设计相一致,比如美国从2016年以来针对非小细胞肺癌的多款药物的适应症批准都是基于OS数据或OS数据加上PFS数据,如果是仅有PFS数据的药物也不符合美国的临床终点的实际情况。 另一方面,对照组的设计也需要符合美国临床实际,即需要将该疾病领域已经获批的标准疗法作为对照组,而不能简单的以安慰剂作为对照组。这对临床试验设计提出了更高的要求,因为获得上市批准的药品是不断增多的,不断会有新的药品获批并成为标准疗法。而临床试验,尤其是以OS为终点的临床试验往往会持续较长时间,这要求企业在进行临床试验设计的时候,提前考虑到未来几年的新药格局及标准疗法的变化,提前选择当时可能尚未获批或尚未进入指南的药物。特定情况下可以考虑灵活的监管政策。对于几种特殊情况,包括针对为满足的临床需求的药物,针对罕见病的药物(罕见病的MRCT难度较大),针对新的药物种类,FDA会根据监管规则,灵活审评临床数据。 值得注意的是,君实的特瑞普利单抗在美国申请上市的适应症是鼻咽癌,而鼻咽癌在美国发病率低,属于罕见病。因此,尽管特瑞普利单抗完成的两项鼻咽癌注册临床没有包含美国人,但可能是适用于灵活监管政策的情况,因此在君实收到的FDA回复中,并没有建议另外完成一项国际多中心的注册临床,而仅仅是需要变更质控流程。
国产创新药美国申报详情 基于以上启发,我们梳理了目前在美国已经提交了上市申请的药物的关键信息。目前已经提交美国上市申请,还在等待审评结果的国产创新药包括百济的替雷利珠单抗,亿帆医药的贝格司亭,以及百济泽布替尼的新增适应症。 目前,亿帆医药贝格司亭的审批结论时间已经由于疫情原因推迟,百济的两个申请的审批结论时间分别是今年7月和10月,从这三个药物的支持上市申报的临床试验来看,都符合国际多中心或美国临床的要求,满足人群的适用性。我们建议2022年下半年密切关注创新药出海获批的进展。此外,2022年5月12日,恒瑞医药公布其PD-1卡瑞利珠单抗联合阿帕替尼一线治疗晚期肝细胞癌的临床试验达到主要临床终点。这项临床同样是国际多中心III期临床,以索拉非尼作为对照药物,并且达到了PFS和OS的双终点。恒瑞医药计划近期向FDA提交沟通交流申请。 我们认为,各个疾病领域标准治疗药物的不断进化和演变,尤其是发达市场的药物进展往往快于国内,以及FDA等监管审批体系和要求的不断变化,对于国内创新药出海提出了更高的要求。国内创新药需要完成大型的国际多中心临床试验,需要选择推荐药物甚至提前选择未来的推荐药物作为对照,并且达到OS获益的临床终点金标准。或者是差异化的在海外药物尚未涉及的、未满足临床需求较大的罕见疾病领域做出明确疗效数据。这对于国内药企的创新药研发能力、临床试验设计能力、国内外临床试验推进能力、以及与监管的沟通能力都有全方位的要求。国产创新药走向世界目前还处于探索阶段,但是随着国内各类创新药企业在探索中坚持前行,预计不远的将来我们即将迎来创新药出海的全面开花结果。
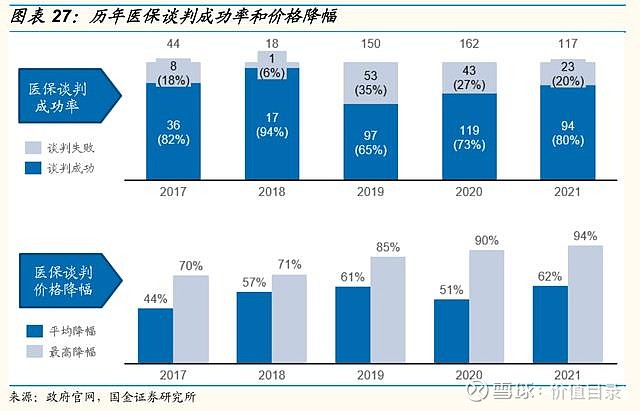
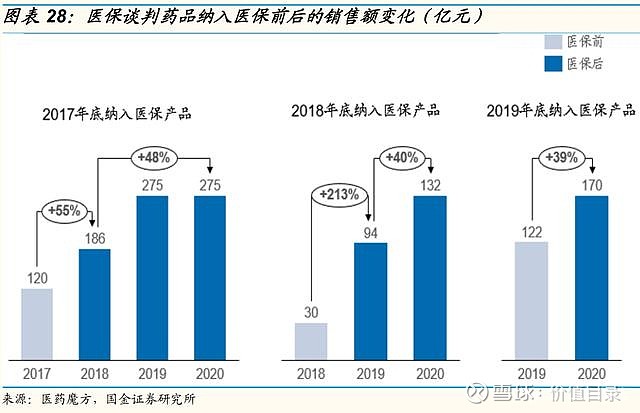
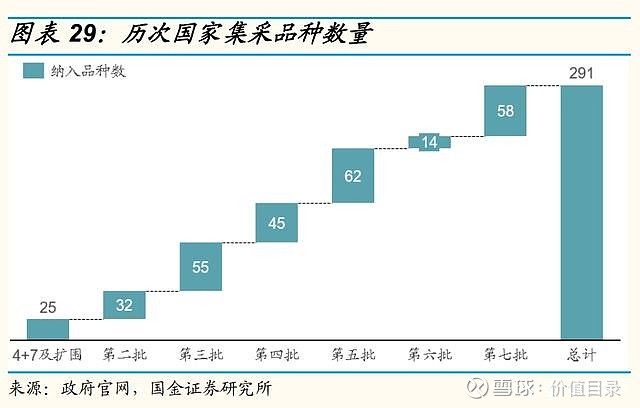
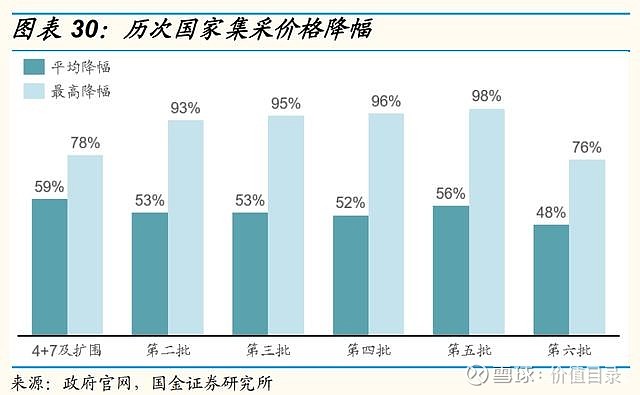
医保谈判价格降幅稳定,助力创新药放量 中国医疗保险制度逐渐完善,2018年国家医疗保障局正式成立,组织制定城乡统一的药品、医用耗材、医疗服务项目、医疗服务设施等医保目录和支付标准,建立动态调整机制,制定医保目录准入谈判规则并组织实施。其中,医保目录调整由原来的多年调整一次,逐步趋向每年常态化调整,2020年起,医保动态调整机制开始实施。近年来,医保谈判成功率较高。2017年是第一次医保谈判,2018年是肿瘤药专项医保谈判,整体来看,2017年至2018年医保谈判成功率较高,但涉及药品数量较少。2019年至2021年,参与医保谈判的药品数量显著上升,医保谈判成功率也由2019年的64.7%逐渐上升到2021年的80.3%,每年医保谈判成功的药品数量分别为97/119/94个(含目录内谈判成功品种),较2017年至2018年有大幅上升。新增品种的价格降幅趋于稳定,平均降价幅度保持在50%至60%左右。 尽管医保谈判降价幅度较高,但从过去几年医保谈判的产品来看,总体都实现了量价交换,达到了销售额的显著增长。 我们认为,进入医保目录是创新药产品实现放量和销售额增长的重要和有效的关键里程碑事件。随着医保动态调整制度的日益完善,以及企业对医保谈判规则的逐渐熟悉,创新药产品有望在上市后一年之内进入医保目录,我们看好创新药通过医保谈判实现加速放量。 集采常态化、制度化开展,价格降幅趋于稳定 自2018年的“4+7”试点城市药品集中采购,国家组织药品集采已经完成了6批,平均降幅均在50%左右。2021年11月,第六批国家胰岛素专项集采开标,首次将生物药纳入国家药品集采范围。国家前六批集采总共涉及234个品种,在集采之前这些品种的市场规模总量约为1500亿元。2022年1月,第七批集采报量正式启动,涉及58个品种。
2021年底至2022年初,距离第一批集采执行时间已经过去两年,距离第三批集采执行过去一年时间,不少集采约定期满的产品面临集采续约。国家各项政策文件强调保证中选药品长期稳定供应,确保中选产品降价不降质的要求。吉林省第一批第三批集采到期竞价续约中,部分产品中标价也出现了6%-126%的提价。陕西医保局发布的《关于做好国家组织药品集中带量采购协议期满后续工作的函》中也指出,允许中选产品续约时价格适当上浮。续约允许提价的机制有利于企业保留合理利润空间,维持长期稳定供货。 我们认为,一方面随着大品种率先集采,后续集采的品种规模会逐渐缩小,集采对仿制药存量企业的影响也会逐渐减弱;另一方面随着集采降价温和及续约允许提价的边际利好,仿制药业务仍能带来合理的利润空间。 我们看好2022年下半年医药政策边际回暖和长期创新驱动下的中国创新药投资机会。我们看好具有国际化能力的头部biotech企业,百济神州已有海外上市销售泽布替尼的经验,PD-1特雷利珠单抗也即将迎来FDA的审批结果;君实生物通过海外合作伙伴,已经有新冠中和抗体在海外获批EUA并销售,看特瑞普利单抗解决变更质控流程的问题后,有望今年重新提交BLA申请并相对快速的获批。 我们看好具有国际化能力的头部大型药企,恒瑞医药的卡瑞利珠单抗联合阿帕替尼对比索拉非尼一线治疗晚期肝癌的国际多中心III期临床试验已经达到PFS和OS双终点,计划近期提交与FDA的上市交流申请。我们看好具有差异化创新能力与全球前沿技术的biotech企业,金斯瑞生物科技的子公司传奇生物的CAR-T细胞疗法的首次获批上市,距离全球首款CAR-T细胞疗法上市不到4年时间,荣昌生物的HER2ADC维迪西妥单抗具有差异化竞争力,通过与ADC领域领先企业Seagen的合作,有望充分挖掘产品潜力,实现海外市场上市销售。
中国创新药研发逐步与国际接轨。2021年中国获批上市的创新药数量和种类分布与美国相当,疾病领域分布与美国类似。国内创新药审批流程日益完善,优先审评、附条件批准、突破性疗法以及境外上市的临床急需新药四条加速审批路径日益成熟;创新药中位审批时间从2016年的接近800天缩短至2021年的400天左右;同靶点创新药在中美获批的时间差也呈逐渐缩短;多方面数据表明,中国创新药研发正在逐步与国际接轨。 国内外合作愈发频繁紧密。近年来,中国药企与海外市场合作交易频发,无论是中国药企引进海外创新药产品交易数量,还是中国国产创新药的海外市场权益的对外授权交易数量,都呈快速增长趋势,这首先表明中国药企的合作意识和合作落地的能力都在增强。2021年不少国内药企在一年之内引进了3项或3项以上产品,也有不少金额较大的大型交易。产品引进一方面能快速补充企业的产品管线,另一方面也能通过合作加速中国创新研发与国际领先水平的接轨。2021年以来,几项大金额的对外授权交易,尤其是百济神州与诺华、君实生物与Coherus的二度合作,一方面为国内企业迅速补充现金流,另一方面也反映出中国药企的研发实力逐步受到海外药企的认可。 国产创新药出海在探索中前行。发达市场的药物研发不断进展、监管审批要求不断变化,对于国内药企的创新药研发能力、临床试验设计能力、国内外临床试验推进能力、以及与监管的沟通能力提出了全方位的更高的要求。百济神州的泽布替尼和传奇生物的cilte-cel顺利获得FDA批准,更多提交上市申请的产品尚未获批,但也给我们带来了宝贵的经验。国产创新药出海走向世界目前还处于探索阶段,但是随着国内各类创新药企业在探索中坚持前行,预计不远的将来我们即将迎来创新药出海的全面开花结果。 医药政策边际回暖趋势。进入医保目录是创新药产品实现放量和销售额增长有效手段。随着医保动态调整制度的日益完善,创新药产品有望在上市后一年之内进入医保目录,看好创新药通过医保谈判实现加速放量。仿制药集采常态化制度化开展,后续集采产品规模缩小对仿制药存量企业的影响逐渐减弱,随着集采降价幅度稳定及续约允许提价的边际利好,仿制药业务仍能带来合理的利润空间。我们看好2022年下半年医药政策边际回暖和长期创新驱动下的中国创新药投资机会。 $恒瑞医药(SH600276)$ $君实生物-U(SH688180)$ $百济神州-U(SH688235)$ 以上为报告节选,报告原文: 医药生物-中国医药研发创新专题(2021):在探索中坚持前行-国金证券[王班]-20220523【30页】 访问【价值目录】WEB端,可以下载相关研报 |
【本文地址】