复旦大学张俊良、杨俊锋教授团队Angew. Chem.:钯催化的次磺酰胺不对称芳基化构建硫亚胺 |
您所在的位置:网站首页 › 张俊才教授政治 › 复旦大学张俊良、杨俊锋教授团队Angew. Chem.:钯催化的次磺酰胺不对称芳基化构建硫亚胺 |
复旦大学张俊良、杨俊锋教授团队Angew. Chem.:钯催化的次磺酰胺不对称芳基化构建硫亚胺
|
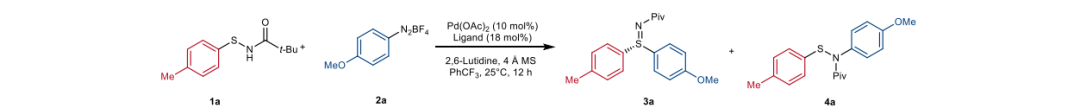
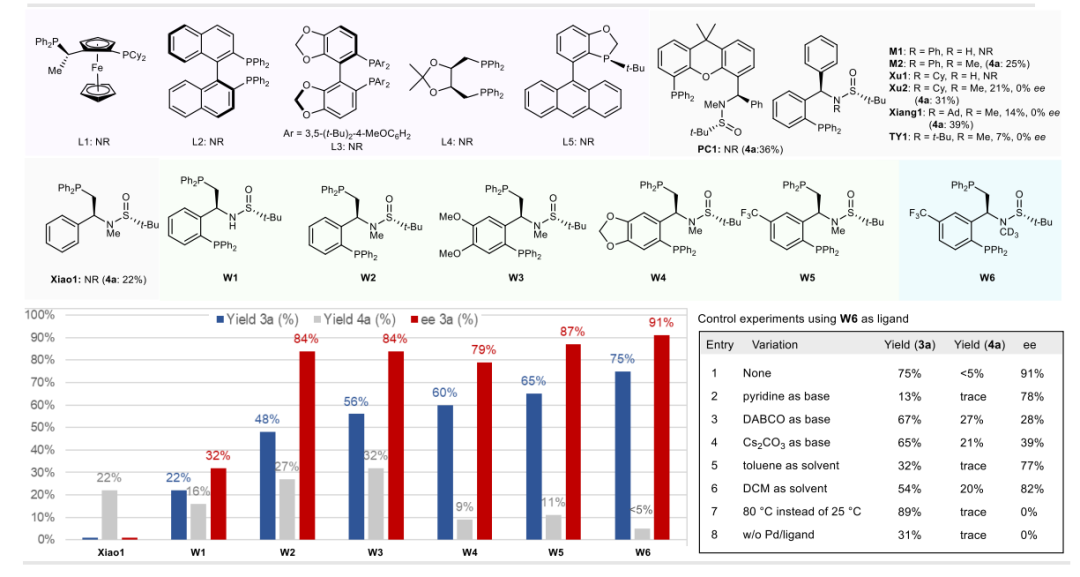
钯催化的次磺酰胺不对称芳基化构建硫亚胺 作者以对甲基次磺酰胺1a与对甲氧基苯基重氮盐2a作为模板底物,以醋酸钯为催化剂,PhCF3作为溶剂,2,6-Lutidine为碱对手性配体进行了条件探索。考虑到双膦配体在C-S交叉偶联中的成功应用,首先作者筛选了一批市售的手性双膦配体,它们都没有产生S-芳基化或N-芳基化产物。接下来,作者筛选了实验室开发的手性亚磷酰胺类配体,这些配体已广泛应用于过渡金属催化的偶联反应。使用M2、PC1和Xiao1与N原子上的甲基进行反应,仅发生了N-芳基化。使用含有电子丰富的P-Cy和P-Ad基团而不是P-Ph基团的配体(M2)进行反应,产生了S-芳基化产物,并伴有一些N-芳基化产物。但令人失望的是,这些配体都没有在S-芳基化中引发任何对映选择性控制。随后当测试双膦配体W1时,获得了22%收率和32% ee的目标S-芳基化产物,同时还有16%的N-芳基化产物。如果在配体的N原子上加入甲基,对映选择性可以显著提高到84%。这一结果与单膦配体如Xiao1的表现形成鲜明对比。因此,作者对W1骨架上的取代基进行了广泛筛选,发现骨架对位含有电子吸引三氟甲基(CF3)取代基的配体(W5)可以将对映选择性提高到88% ee,同时具有更高的S-化学选择性。相反,配体上的电子供给取代基(W3和W4)略微降低了产率和对映选择性。同样,对W5的N原子上的取代基也进行了改造,发现氘代甲基(W6)可以进一步将ee值提高到91%。这可能是由于氘原子的微妙立体效应,次磺酰胺上氮原子的保护基也起到了关键作用,没有氮原子上羰基的底物无法生成产物。温度对对映选择性也有关键影响,因为作者在较高温度下观察到了严重的背景反应。加入分子筛可以略微提高产率。最后,通过进一步筛选碱、溶剂和预催化剂,作者确定了在常温下使用Pd(OAc)2、W6和2,6-Lutidine在PhCF3中的组合为最佳催化体系,产率为75%,对映选择性为91% ee。
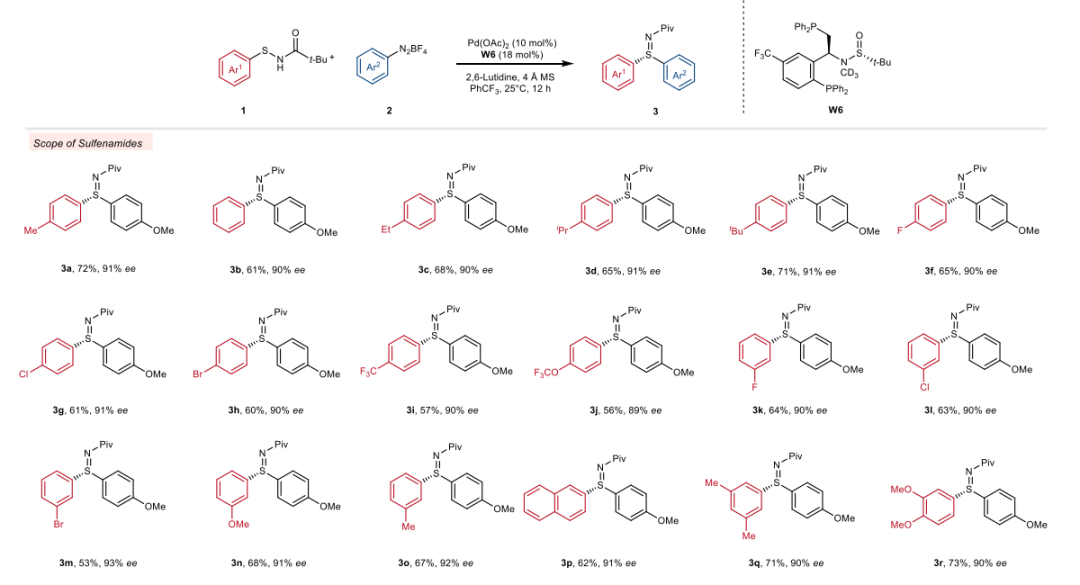
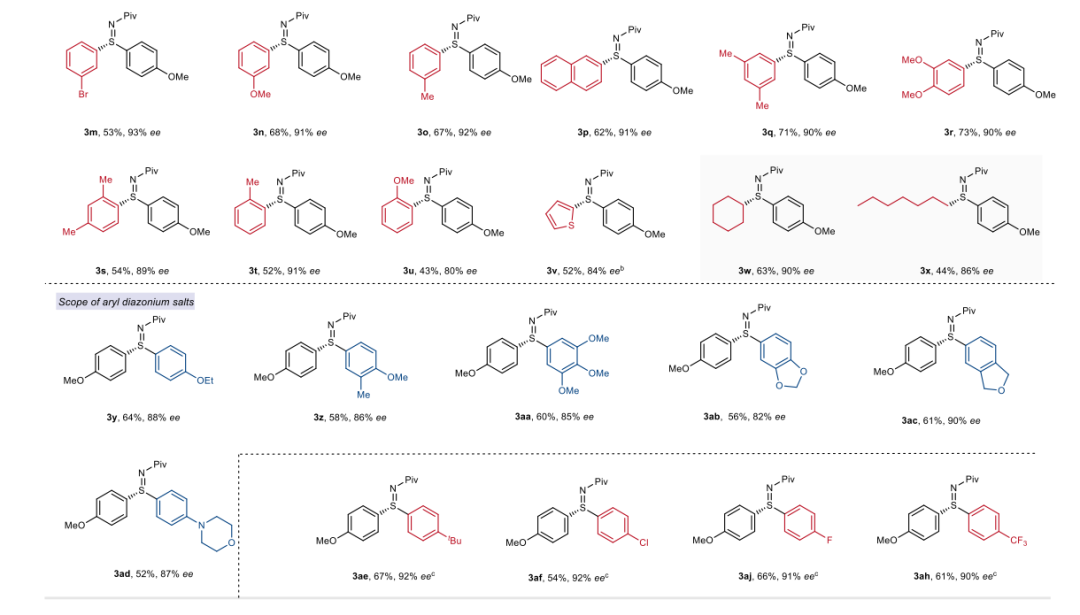
(来源:Angew. Chem.) 在确定了最优反应条件后,作者首先对不同的次磺酰胺进行了考察。给电子和吸电子的芳基次磺酰胺在对位和间位取代基上均能顺利反应,生成高产率和高对映选择性的硫亚胺产物(3a至3r,89至92% ee)。如烷基(3b-3e, 3o, 3q)、氟(3f)、三氟甲基(3i)、三氟甲氧基(3j)、甲氧基(3r)均与S-芳基化反应兼容。具有挑战性的邻位取代基也可以反应(3s和3t),而含有配位原子的邻位取代基则导致选择性略低(3u)。杂环如噻吩也能很好地反应,生成52%收率和84%ee的产物3v。值得注意的是,卤素基团如氯和溴ye也能够很好地耐受(3g和3h),这为进一步转化提供了可能性。此外,该方法对烷基取代的次磺酰胺也有效,生成的芳基烷基硫亚胺产物(3w和3x)具有中等收率和良好的对映选择性(86-90% ee)。随后,作者考查了芳基重氮盐的范围,给电子芳基重氮盐是合适的底物,产物具有中等收率和良好的对映选择性(3y至3ad,82至90% ee)。与作者的预期相反,缺电子底物在这种条件下不兼容,生成的产物对映选择性从中等到较差,这可能是由于容易发生背景反应。然而,通过简单交换次磺酰胺和重氮盐的芳香部分,这一限制可以解决,如3ae至3ah所示。当作者尝试将亲电试剂扩展到芳基卤化物,该反应不能发生,未能得到目标产物。值得注意的是,在室温下没有观察到产物的消旋化,表明这些硫亚胺结构具有很高的稳定性。
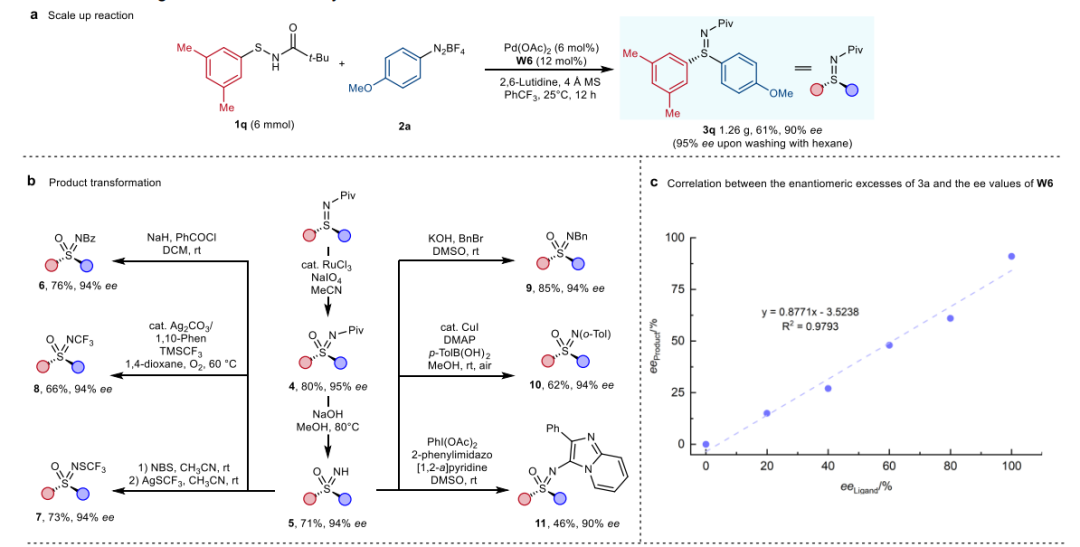
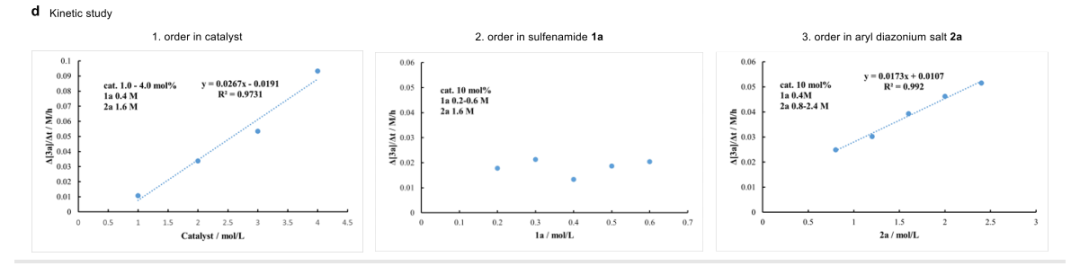
(来源:Angew. Chem.) 在随后的克级规模制备中可维持产率和对映选择性。二芳基硫亚胺3q可以在较低催化剂用量下以61%的收率和90%的对映选择性得到1.26克。在衍生转化中可在构型保持的条件下得到多种六价砜亚胺,证明了其广泛的应用前景。首先,通过RuCl3催化氧化方法进行简单的硫氧化,得到相应的磺酰亚胺4。随后,在碱性水解条件下,N-Piv可以容易地移除,得到71%收率的磺酰亚胺5。用苯甲酰氯或苄基溴处理磺酰亚胺5,可以顺利生成苯甲酰或苄基保护的磺酰亚胺6和9。在银催化剂的存在下,以Me3SiCF3作为三氟甲基化试剂,可以直接形成N-CF3键,生成N-三氟甲基化的磺酰亚胺衍生物7。或者,通过一锅两步法,在磺酰亚胺氮上的溴化,随后与三氟甲基硫代银反应,生成N-SCF3取代的磺酰亚胺8,产率良好。使用典型的Chan-Lam条件,在磺酰亚胺5上引入芳基,也可以得到磺酰亚胺衍生物10。通过PhI(OAc)2氧化咪唑并吡啶化,进一步对磺酰亚胺5进行合成修饰,得到11。值得注意的是,这些合成修饰都很好地保留了硫中心的立体化学完整性。作非线性效应实验发现产物的ee值与配体的ee值之间呈线性关系,表明单体W6-Pd可能是实际的催化物种。在最佳反应条件下的次磺酰胺1a和重氮盐2a之间的动力学分析实验表明,反应速率数据表明钯催化剂和重氮盐2a的浓度呈一级效应,而与次磺酰胺1a的浓度则呈零级效应。这一结果表明,通过氧化加成形成芳基-Pd复合物是速率决定步骤。
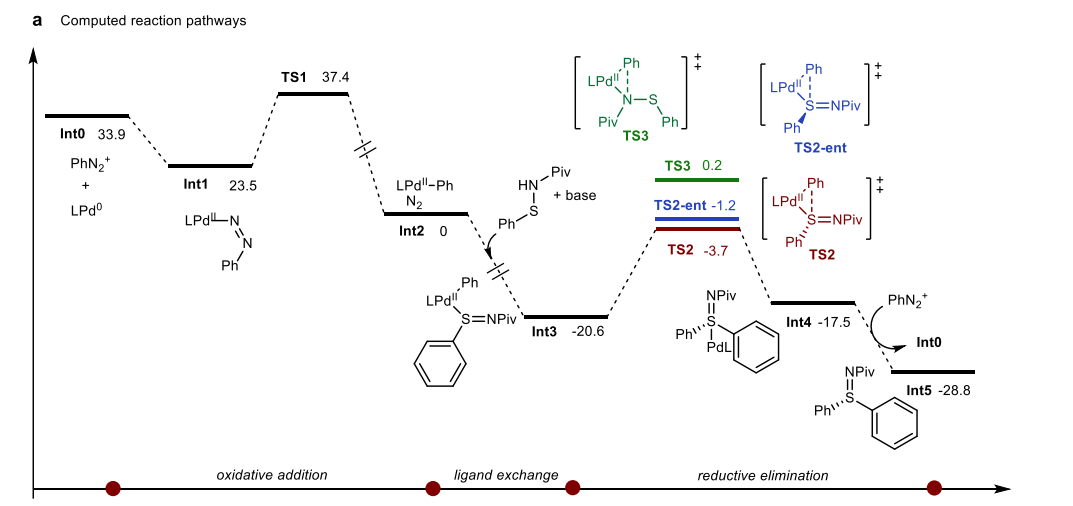
(来源:Angew. Chem.) 为了进一步了解反应机制,作者首先用苯基重氮盐和次磺酰胺1a作为模型反应物,从理论上研究了C-S交叉偶联路径。根据先前关于Pd(0)复合物与芳香重氮盐氧化加成的文献,这一过程通过一个四元环过渡态发生。首先,钯芳基偶氮基团通过Pd(0)复合物与ArN2+的重氮基团氧化加成迅速形成。随后,通过13.9 kcal/mol的能量屏障形成四元过渡态TS1。最后一步是Ar−N2键的断裂,生成芳基-Pd中间体Int2。值得注意的是,这在形式上是一个β-碳消除过程,因此在C-N键断裂期间没有发生氧化态变化。阳离子芳基-Pd中间体Int2在碱存在下与次磺酰胺配位,分别在硫和氮上生成中性芳基-Pd(II)复合物。随后从这个芳基-Pd(II)复合物进行还原消除(TS2、TS2-ent、TS3),生成S-芳基化和N-芳基化产物,同时释放Pd(0)催化剂。因此,反应的选择性由还原消除步骤决定,S-芳基化的TS2相比N-芳基化的TS3显著有利(ΔΔG = 3.9 kcal/mol)。此外,作者发现两个非对映过渡态TS2和TS2-ent之间的自由能差异为2.5 kcal/mol,与观察到的对映选择性一致。同时,作者还推测了通过六元过渡态进行还原消除的可能性,钯上的S,O-双齿配位。然而,计算结果表明这些双齿配位模式比三元还原消除能量更高。总体而言,能量剖面图表明主要形成(S)构型的硫亚胺产物,这与当前转化的对映选择性和化学选择性实验观察结果一致。 随后,通过对立体环境的分析研究了化学选择性和对映选择性的起源。虽然TS2的催化剂和底物的扭曲能量(ΔEdis-LPd和ΔEdis-sub)比TS2-ent更强烈,但影响反应的主导因素是催化剂和底物之间的相互作用能量(ΔEint),这导致TS2-ent中更高的焓值。这个结果与在TS2-ent中观察到的更强的C–H···O相互作用(2.14 Å ~ 2.75 Å)一致,该相互作用发生在底物酰胺基团中的氧原子与配体中的C–H键之间。这些弱相互作用也通过独立梯度模型(IGM)观察到。然而,与TS2和TS2-ent不同,影响TS3的主导因素是催化剂和底物的扭曲能量(ΔEdis-LPd和ΔEdis-sub),这表明TS3涉及更多的底物-配体的空间排斥,因为次磺酰胺和苯基取代基之间的距离较近。
(来源:Angew. Chem.) 总结来说,复旦大学张俊良、杨俊锋课题组开发了一种高效的钯催化手性选择性C-S交叉偶次磺酰胺和芳基重氮盐的方法。通过一步,制备了一系列以前难以获得的具有高化学和对映选择性的二芳基硫亚胺和芳基烷基硫亚胺。手性硫亚胺可以作为获得一系列手性S(VI)结构的关键结构,其中手性亚磷酰胺双膦配体对于实现这些高选择性至关重要。通过DFT研究进一步探讨了选择性的起源,结果表明底物羰基和配体之间的弱相互作用可能是导致立体选择性的原因。除了直接的实用性之外,作者期待这项工作能够激发并加速手性选择性C-S交叉偶联转化的研究和发展。 这一研究成果于 2024 年 6 月 27 日以标题“Enantioselective Arylation of Sulfenamides to Access Sulfilimines Enabled by Palladium Catalysis” 在线发表于Angewandte Chemie International Edition (DOI: 10.1002/anie.202409541)。课题组博士生袁茵为该论文第一作者,复旦大学张俊良、杨俊锋教授为该论文共同通讯作者。 |

【本文地址】