罕见病诊疗指南(2019年版)之进行性肌营养不良 |
您所在的位置:网站首页 › 基因dmd › 罕见病诊疗指南(2019年版)之进行性肌营养不良 |
罕见病诊疗指南(2019年版)之进行性肌营养不良
|
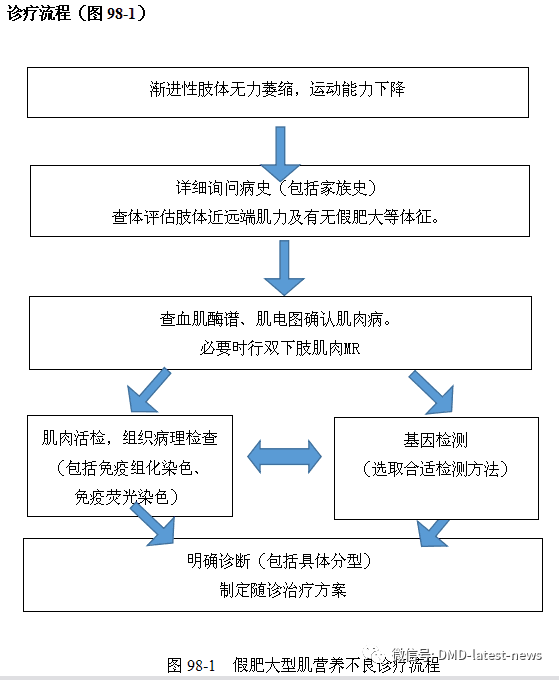
Duchenne/Becker型肌营养不良相关辅助检查如下: 1.血清学检测肌酸激酶(CK)、乳酸脱氢酶(LDH)、羟丁酸脱氢酶(HBD)、谷草转氨酶(AST)、谷丙转氨酶(ALT)、肌红蛋白(Myo)在肌细胞损害时释放入血,从而引起血中浓度明显升高,可达正常上限的20~200倍。 2.肌电图肌电图对于判定肌肉疾病很重要,尤其在病情尚不明显,特别是轻型BMD肌酶升高不突出时。需通过针极肌电图配合神经传导速度检查,确定为肌源性损害。幼龄早期BMD患儿在检查配合不佳情况下,可能无特殊发现。 3.肌肉MR在肌肉病变发展的不同阶段,通过肌肉MR可发现肌肉组织中存在炎性水肿和(或)脂肪替代,同时帮助明确受累肌群分布特点和病变程度。可用于辅助诊断和随诊病情进展。 4.肌肉活检进行性肌营养不良患者肌肉组织呈肌营养不良样形态学改变。通过免疫组化或免疫荧光染色,可以发现肌膜上的Dystrophin蛋白表达完全或部分缺失。另外,肌肉活检还可鉴别炎性肌病、代谢性肌病等。 5.基因检测基因检测对DMD/BMD诊断具有重要价值。基因检测有多种不同方法,疑诊DMD/BMD,一般先用多重连接探针扩增技术(MLPA)检测DMD基因大片段缺失和重复,如果未发现此类拷贝数异常,再用高通量测序技术(NGS)检测微小突变。随着NGS技术和生物信息学分析的发展,目前已可用高通量测序一步法同时检测拷贝数变化和微小突变。 诊断 幼儿期运动发育轻度迟滞,儿童期(5~6岁)运动能力开始下降,并出现步态异常、跟腱挛缩、腰椎前凸等变化,查体可见明显双腓肠肌假肥大现象。结合血肌酶谱明显升高、肌电图呈肌源性损害,可临床疑诊DMD。确诊需基因检测发现DMD基因致病性缺陷或肌肉活检发现Dystrophin蛋白异常。 鉴别诊断 能够引起DMD/BMD类似临床表现的其他神经肌肉病主要包括运动神经元病,如脊肌萎缩症以及其他肌肉病,如其他肌营养不良、炎性肌病、代谢性肌病等。需重点鉴别的疾病如下: 1.其他类型肌营养不良能够引起肌酶明显升高并有假肥大体征的其他类型肌营养不良,包括肌聚糖蛋白病(sarcoglycanopathy)、肢带型肌营养不良2I、肢带型肌营养不良2B、肢带型肌营养不良2A等。这些类型肌营养不良肌酸激酶可数十倍升高,但起病年龄通常更晚,进展更慢,需与Becker型肌营养不良重点鉴别。需要注意的是,肌聚糖蛋白病也可儿童期起病,10余岁丧失行走能力,特别是女性患儿需要重点鉴别。上述肌营养不良均为常染色体隐性遗传且涉及多个基因,一般采用高通量测序(NGS)一次性检测多种致病基因,提高诊断效率。而对于面肩肱型肌营养不良,常规基因检测方法均无法确定4号染色体亚端粒区的结构变化,只能通过经典Southern blot或第三代光学图谱技术(Bionano)等特殊方法确诊。 2.脊肌萎缩症(SMA)根据病情轻重分为多种类型,可新生儿至成人期起病。以肢体近端无力萎缩为主要表现。血肌酶轻度升高或正常。肌电图可见广泛神经源性损害。 3.炎性肌病包括多发性肌炎、皮肌炎、包涵体肌炎等,在各个年龄均可发病,通常起病较急、进展较快。血肌酶谱升高明显。肌电图提示肌源性损害,通常合并大量自发电位等活跃期表现。肌肉活检可见肌肉组织炎性细胞浸润及肌纤维膜MHC-Ⅰ表达增强。值得注意的是,部分类型肌营养不良,如DMD,LGMD2B,面肩肱型肌营养不良等,肌肉活检亦可见明显的炎性反应。 4.代谢性肌病包括糖原累积性肌病、脂质沉积性肌病、线粒体病等,通常呈波动性病程,发病期常快速进展,血肌酶升高。肌肉活检可见肌细胞内糖原沉积、脂滴增多或破碎红纤维等特征性改变。 治疗 Duchenne型肌营养不良目前尚无治愈方法。但通过规范药物治疗、康复训练、定期随诊评估相关系统受累并给予治疗,能够明显延缓疾病进展,延长生存期,提高生活质量。治疗原则如下: 1.药物治疗对于确诊的DMD患者,建议在3岁后、运动功能下降前开始规范口服激素治疗。一般推荐每日疗法,每日口服泼尼松或泼尼松龙0.75mg/(kg·d),早饭后一次顿服。根据治疗反应和副作用情况,周末疗法(周末2天服用整周剂量),间断疗法(服药10天停药10天,疗效弱于每日疗法)是备选方案。口服激素选择上,甲泼尼龙[0.6mg/(kg·d)]和地夫可特[0.9mg/(kg·d),我国未上市]也可考虑,治疗作用基本相当,副作用与泼尼松有所不同。BMD患者病情较轻,一般不长期应用口服激素治疗。 2.康复治疗对于Duchenne/Becker型肌营养不良,规范的家庭康复治疗非常重要。建议确诊后早期、规律开展。应在有相关疾病治疗经验的康复科医师指导下长期坚持,能够延缓疾病造成的关节挛缩、姿势异常等,并能在肌力不足的情况下,维持更好的生活功能和姿态。一些器械康复,如站斜板、足部矫形支具、站立架等也在疾病不同阶段康复中有重要作用。 3.外科手术治疗对于疾病发展过程中出现的脊柱侧弯、关节挛缩等,应行外科评估。矫形手术能够纠正脊柱、关节的结构畸形,有助于维持运动机能和保持呼吸功能。 4.多学科联合诊治Duchenne/Becker型肌营养不良发展过程中造成多器官系统受累,需要多科协作,联合诊治。出现明显骨质疏松后,需在内分泌科指导下,给予二磷酸盐等药物治疗。出现扩张性心肌病,心功能下降后,需在心内科诊治,给予抗心衰药物治疗。出现呼吸功能下降后,需在呼吸科诊治,必要时应用呼吸机辅助呼吸。其他营养、消化、心理等问题,均需在相应科室评估治疗。 5.患者管理和宣教宣教“与疾病共存”理念,患者及家庭需对所患疾病有正确认识,学会自我管理,重视患者及家庭成员的心理健康。 6.新兴治疗随着基因治疗、细胞治疗的快速发展,Duchenne型肌营养不良已有新的基因治疗药物,如外显子51跳跃药物eteplirsen已被FDA批准上市。适用范围更广的AAV载体导入外源截短dystrophin蛋白的基因治疗临床试验正在开展。其他如基因编辑、干细胞治疗、其他调整药物等也都在快速推进中,相信在不远的未来就会出现更多更好的治疗选择,造福于病人。
参考文献 [1] Ke Q, Zhao ZY, Griggs R, et al. Newborn screening for Duchenne muscular dystrophy in China: follow-up diagnosis and subsequent treatment. World J Pediatr,2017, 13(3):197-201. [2] 中华医学会神经病学分会.中国假肥大型肌营养不良症诊治指南. 中华神经科杂志,2016, 49(1): 17-20. [3] Wang CH, Bonnemann CG, Rutkowski A, et al. Consensus statement on standard of care for congenital muscular dystrophies. J Child Neurol, 2010, 25(12):1559-1581. [4] Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol, 2018, 17(3): 251-267. [5] Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol, 2018, 17(4):347-361.返回搜狐,查看更多 |
【本文地址】
今日新闻 |
推荐新闻 |