区分肿瘤细胞的单细胞CNV分析原理 |
您所在的位置:网站首页 › 单细胞基因分析 › 区分肿瘤细胞的单细胞CNV分析原理 |
区分肿瘤细胞的单细胞CNV分析原理
|
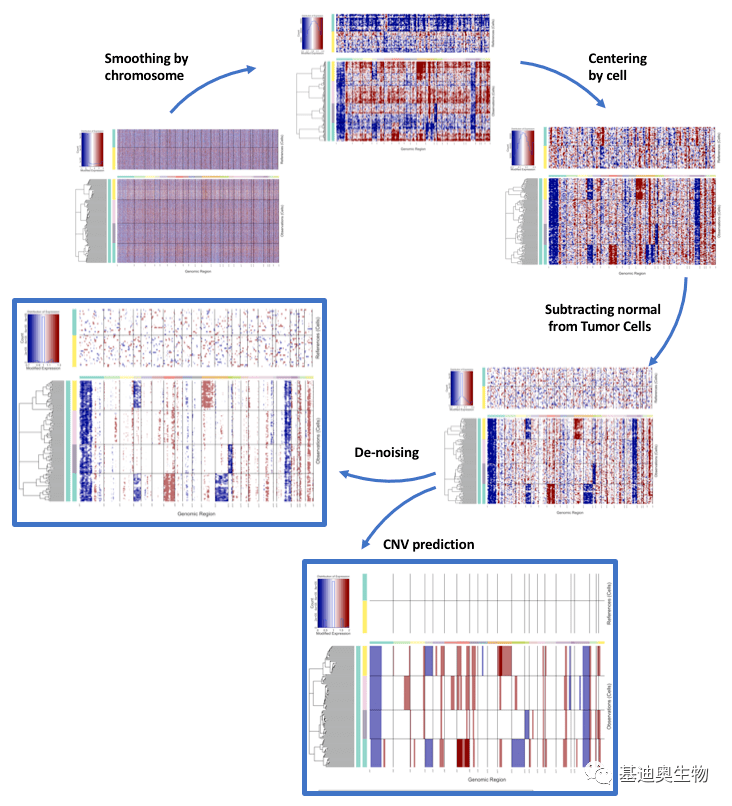
inferCNV算法的具体步骤如下: (1)过滤基因:基因在全部细胞中低表达的基因过滤。 (2)表达量矩阵标准化:为了消除测序深度不同带来的基因表达变化。 (3)肿瘤与参考细胞计算差值:从所有细胞中减去每个基因在参考细胞中对应基因的平均值。由于这种减法是在对数中计算的,所以差值就是相对于正常细胞平均值的对数倍变化值,并且对数倍变化值是有阈值范围的。 (4)染色体水平的滑动窗口:对于每个细胞,沿着每条染色体排列的101个基因使用加权平均值来表示滑动窗口的表达强度。 (5)参考细胞表达强度居中的调整:假设大多数基因不在CNV区域,那参考细胞的每个细胞都可以居中,从而调整表达强度为0。 (6)相对于参考细胞的再调整:再一次从肿瘤细胞中减去参考细胞的平均值,进一步补偿平滑过程后产生的差异。 (7)还原对数变换:这一步会使得重复或缺失的证据在平均值周围更加对称。
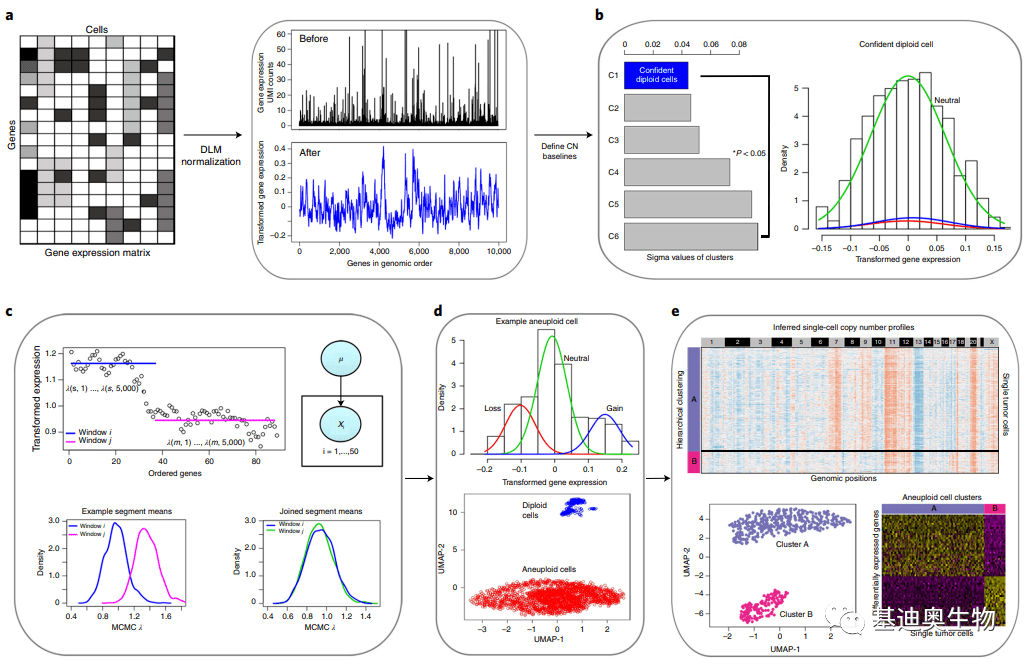
图1 inferCNV运行的步骤 3. copyKAT算法原理[4] copyKAT 通过结合贝叶斯方法与层次聚类来计算单个细胞的基因组拷贝数分布,并定义出亚克隆结构。软件以基因表达矩阵为输入。 (1)首先根据基因组坐标对基因进行排序,并使用UMI做标准化并稳定其方差,同时对单细胞 UMI 计数中的异常值做平滑矫正。 (2)找出正常二倍体细胞子集:通过聚类将细胞分为几个细胞群,然后计算每个细胞群的方差。通过严格的分类标准,具有最小方差的细胞群被定义为“高置信的二倍体细胞”。
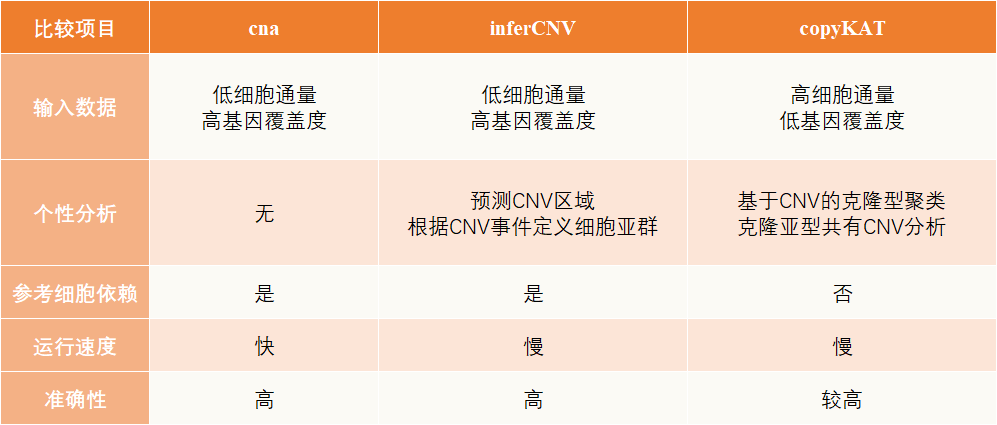
图2 copyKAT分析流程 二、3种单细胞CNV分析软件比较
总结: 我们在执行实际的单细胞转录组肿瘤项目时,想要鉴定TME中的肿瘤细胞可以同时结合三种软件的结果来判断肿瘤细胞,因为对于未知的数据,三种软件的结果可靠性都值得推敲。接着可以根据CNV数据数谱进行肿瘤克隆细胞亚群的聚类,分析肿瘤细胞内部的异质性,也可以鉴定克隆亚群共有的CNV片段上的共有基因,从而分析共有基因在不同亚群的表达量差异,进一步从基因组CNV变异的角度揭示肿瘤发生的机理特征。 具体的单细胞CNV分析内容及应用思路请看下一期介绍。 从单细胞测序概念、数据挖掘、图表可视化到个性化高级分析,系统学习单细胞组学就在基迪奥单细胞线上培训班↓↓↓ 课程信息 时间:2023年4月17-21日 地点:腾讯会议 线上直播授课 培训费用:2200元/人/期 团报福利: 2人组团 2000元/人/期 3人及以上 1800元/人/期 报名方式 方式一: 方式二: 发送姓名、单位、电话到邮箱[email protected],主题注明“单细胞培训班” 方式三: 参考文献 [1] Anoop P. Patel, Itay Tirosh, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014 Jun 20: 1396-1401 [2] Puram SV, Tirosh I, et al. Single-Cell Tranomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell. 2017 Dec 14;171(7):1611-1624.e24. [3] inferCNV:https://github.com/broadinstitute/inferCNV [4] Gao, R., Bai, S., Henderson, Y.C. et al. Delineating copy number and clonal substructure in human tumors from single-cell tranomes. Nat Biotechnol 39, 599–608 (2021). *未经许可,不得以任何方式复制或抄袭本篇文章之部分或全部内容。版权所有,侵权必究。 基迪奥生物|专业定制测序服务 联系方式:020-39341079;[email protected]返回搜狐,查看更多 |
【本文地址】
今日新闻 |
推荐新闻 |