临床试验中药物安全性的统计学考虑 |
您所在的位置:网站首页 › 单位大事件记录格式 › 临床试验中药物安全性的统计学考虑 |
临床试验中药物安全性的统计学考虑
|
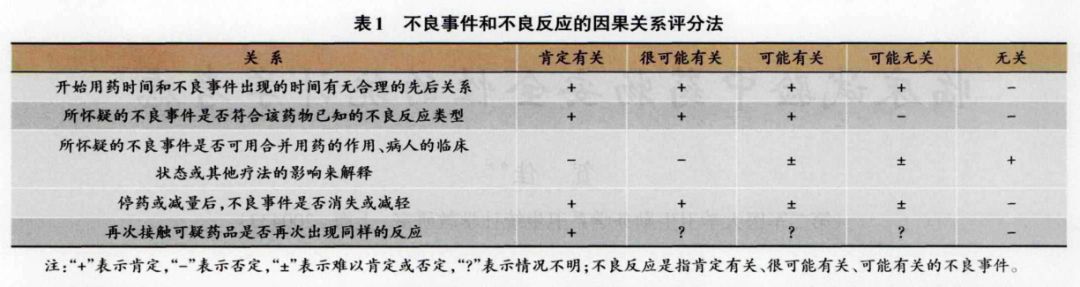
1.2 不良件与下良反应的统计评价 AE是指服用药物的患者或临床试验受试者使用药物后出现的任何不良医学事件。ADR是指在新药或药物新用途的临床试验中,特别是其治疗剂量尚未确定时,与任何剂量的药物使用有果关系的、非预期的有害反应。AE和ADR评价主要采用因果关系分法(表1)描述了5级关系,3个“有关”和2个“无关”,与之有关的均为不良反应。
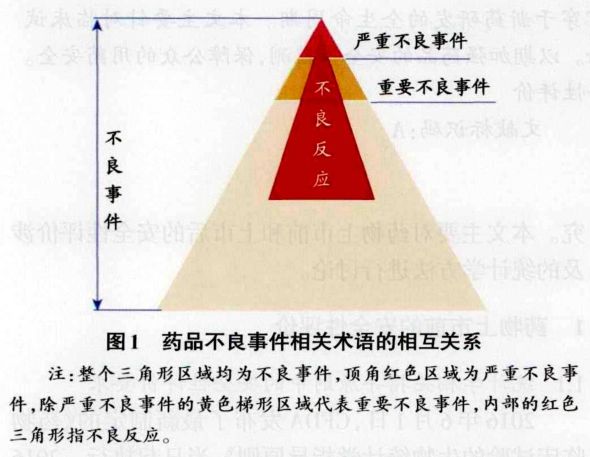
严重不良事件(Serious Adverse Event,SAE)是指在临床试验过程中,在任何剂量时患者或临床试验受试者发生的不可预见的以下临床事件:死亡、危及生命、需要住院治疗或延长目前的住院治疗时问,导致永久性或显著的功能丧失或残废、导致先天性畸形或出生缺陷的重要的医学事件(Important Medical Events)。 重要不良事件(Significant Adverse Event)指的是严重不良事件外,发生的任何导致采用针对性医疗措施(如停药、降低剂量和对症治疗)的不良事件和血液学或其他实验室检查明显异常(图1)。
1.3 统计学相关指导原则中的安全性分析要求 安全性分析数据集(SafetySet,SS)是指至少接受过—次治疗且有安全性评价的受试者。统计学分析是根据事先确定的统计分析计划,采用描述性统计分析方法,必要时辅以P值和置信区间,需要注意的是安全性评价指标需采用统—的不良事件编码词典,如MedDRA等。统计学分析的内容如表2所示。
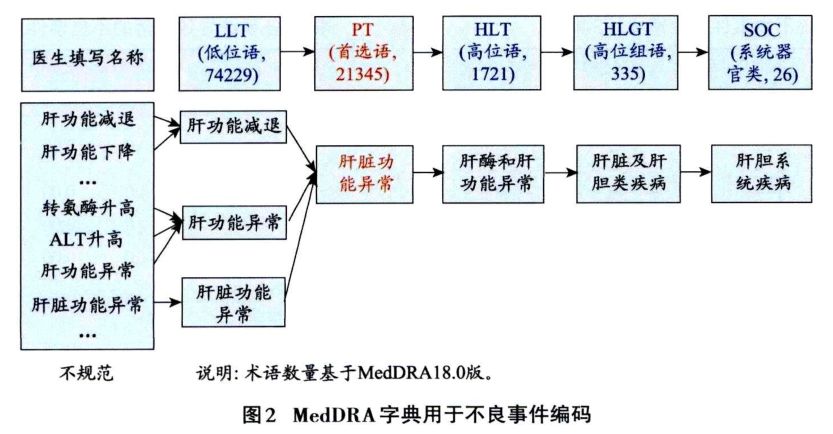
1.3.1 不良事件的编码 编码就是对特定对象或事物进行分类的过程。即按照标准字典或术语集进行分类,是对事物的多方面性质的解释,根据编码可以方便对不良事件进行统计分析。不良事件编码词典如MedDRA、WHO-ART,药物编码如WHO Drug,疾病编码如ICD-10等。不良事件编码以MedDRA字典为例(图2),不同医生对于肝功能变化有各种描述,为了方便统计,首先将各种肝功能描述用低位语进行编码,可归为肝功能减退、肝功能异常和肝脏功能异常;然后进行首选语和高位语的归类,最后到系统器官类归为肝胆系统疾病。
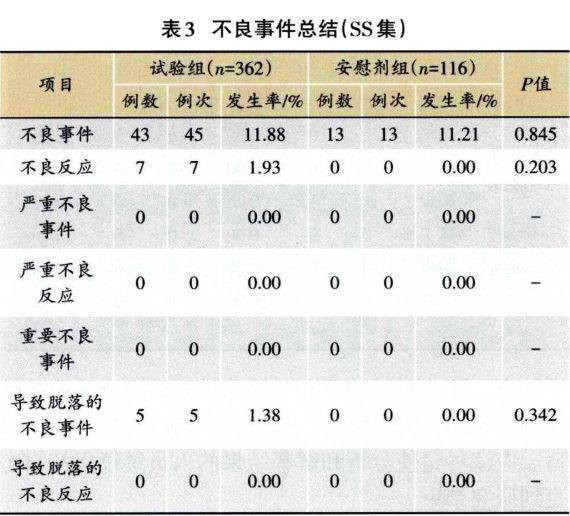
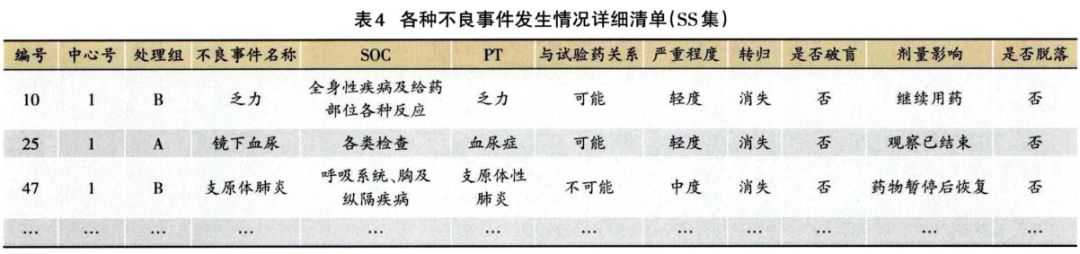
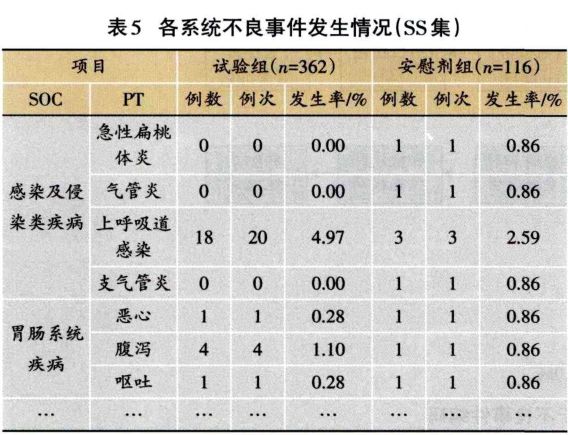
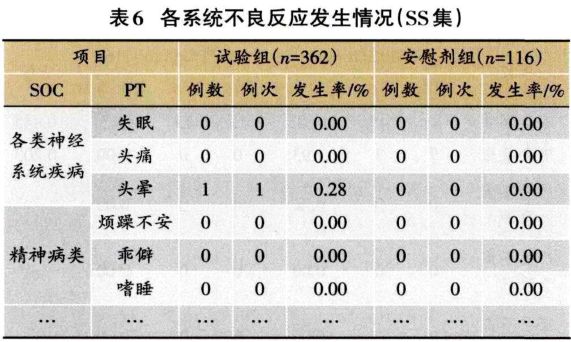
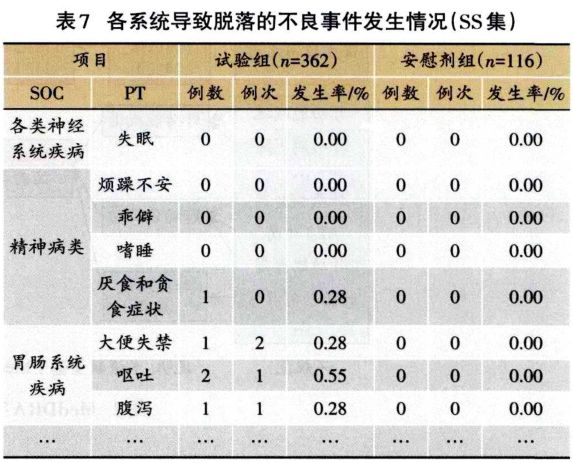
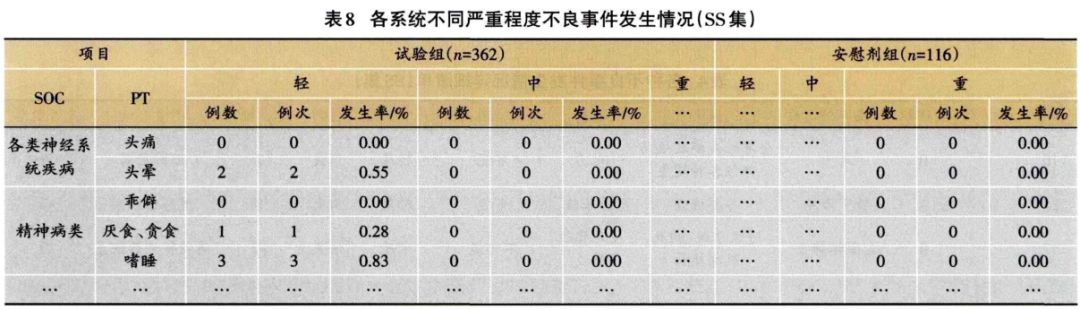
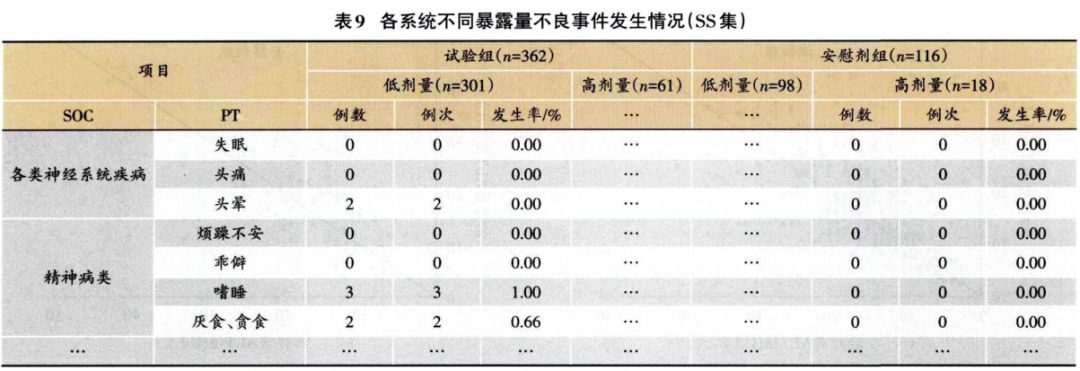
1.3.2 安全性分析实例 临床试验的题目为“XX中药治疗XX病的安全性和疗效”,采用安慰剂对照,样本量480例,试验组和安慰剂组患者比例为3:1:。首先,将不良事件、不良反应、严重不良事件和反应、重要不良事件等项目列出(表3)。然后,将各种不良事件的MedDRA编码详细内容和关系列出(表4);此外,按照各个系统不良事件、不良反应的发生进行列表,按照MedDRA编码后的SOC和PT名称归类,将对应的例数、例次和发生率列出(表5、表6);导致脱落的不良事件和各系统不同严重程度的不良事件也按照通样方法列出(表7、表8)。另外,不同暴露剂量的不良事件发生情况(表9)和依从件均列出清单。
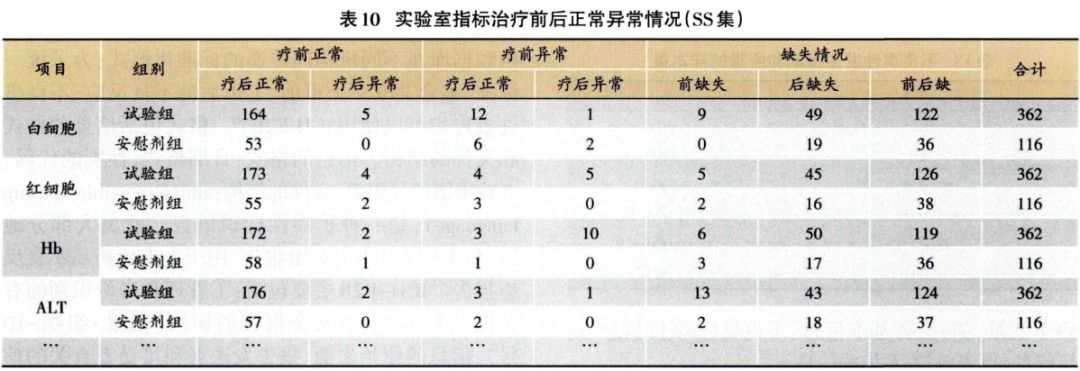
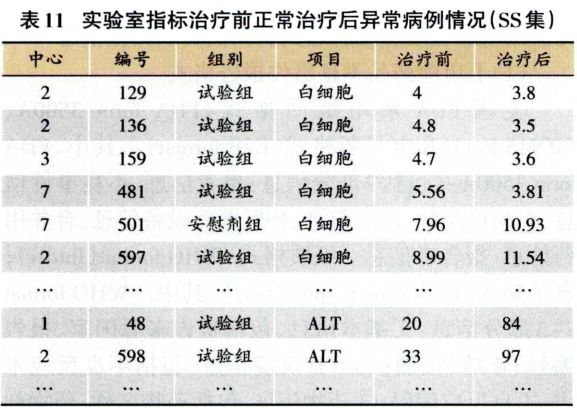
实验检查指标的分析,重点关注治疗前后实验室指标临床意义变化的交叉表(表10)。由表10可见,主要关注治疗前正常但治疗后异常这—列数据;其次关注治疗前常异常和治疗后异常—列,查看患者在治疗后是否病情更严重、再观察具体指标值,如表11所示,逐例审核数值的变化情况。
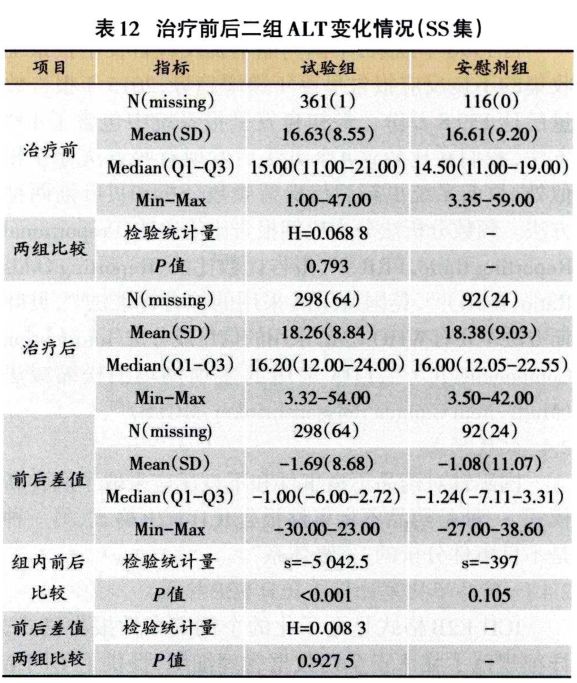
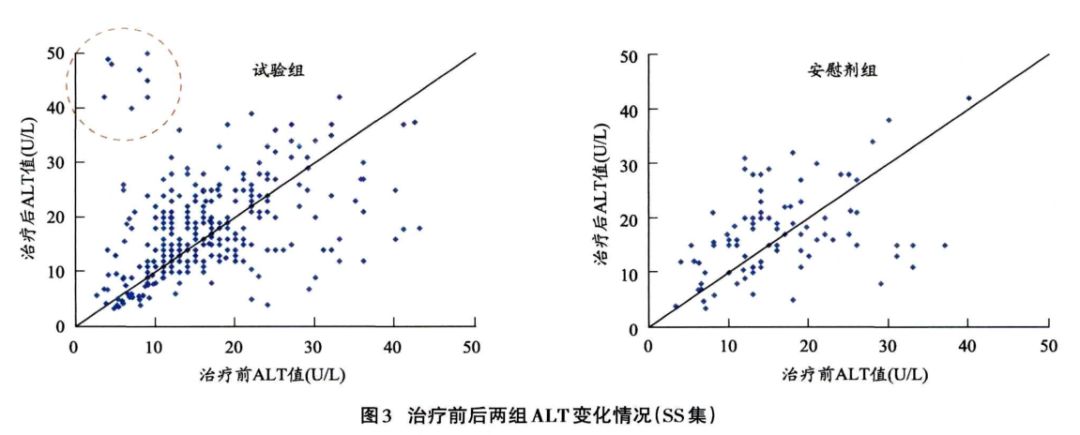
采集的实验室指标还可以进行数值的描述性分析,进—步观察治疗前后试验组和安慰剂组ALT是否整体升高,详见表12。此外还可以通过绘制散点图清晰展示每位患者治疗前后ALT、的变化情况,如图3所示,试验组有个别病例远离斜线,分布在斜线左上方,提示治疗ALT升高;而安慰剂组分布较均匀。
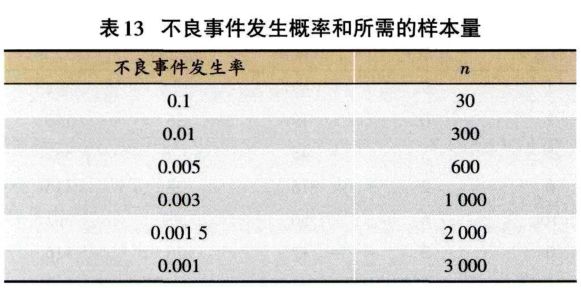
1.4 注意事项 安全性研究与有效性研究—样,也需要考虑随机化、对照与盲法、样本量等问题,还要关注安全性事件的破盲问题。 1.4.1 对样本量的考虑 I期、Il期、III期临床试验—般不会因为安全性事件而考虑样本量问题。上市前样本量考虑主要参考疗效的样本量和法规。上市后样本量主要根据单组不良事件发生率计算。可以参考表13,如果有5%的发生率,则需要600例样本量。
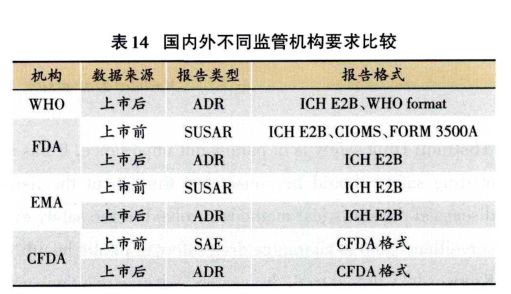
1.4.2 严重不良事件的破盲问题 出现严重不良事件时,无需立即全面破盲。如果破盲结果对受试者将接受的医疗处理没有影响,可以不破盲。除非对试验药和对照药导致的不良反应的处理方法不同,这种情况可以进行个例破盲,切勿全部破盲。应该尽量使分析和解释结果的人员继续保持盲态直到研究结束。 2 药品上市后的安全性评价 药品上市后有主动监测和被动监测,IV期临床试验属于主动监测。上市前的临床试验均为小样本的临床试验,不足以发现更多的不良反应情况;而上市后的大样本临床试验有利于发现—些特殊安伞性问题,如:罕见不良反应、迟发性不良反应、长期的不良反应、发生于特殊人群的不良反应、合并用药出现的不良反应。这就要求进行药物安全警戒(Pharmacovigilance,PV),PV是与发现、评价、理解和预防药品不良反应或其他任何可能与药物有关问题的科学研究与活动,包括主动监测和被动监测。 2.1 主动监删 主动监测是指由主体方(政府、药品生产企业等)针对某—药品,为探索某个或某些安全性问题的性质和/或程度等,基于各种适宜科学方法而展开的各种活动、行为和研究。主动监测能获取人口学资料,药物、诊断信息;获取随访过程中发生的所有不良事件;可以计算药品不良反应发生率。主动监测主要是在上市后进行安全性研究、IV期临床试验;另外还可以进行哨点监测、集中监测计划和处方事件监测。 2.2 被动监测 被动监测是指药品在上市后使用过程中,由医疗卫生专业人员、药品经营、生产者、消费者等将所发现、获知或经历的可能与药品安全有关的信息,上报给药品监管机构、生产企业、医疗卫生专业人员或其他组织的过程。被动检测信息将上报至自发呈报系统(Spontaneous Reporting System,SRS)。该系统目前还存在不能计算药品不良反应发生率和漏报的问题。中国药品不良反心自发呈现系统需要国家中心、省中心授权方可进入。中国药品不良反应自发呈报系统包括69个变量,如:患者基本信息、不良反应/事件信息、药品信息、报告单位/人相关信息。 2.3 统计分析方法 自1998年以来,中国药品不良反应自发呈报系统收集的不良反应报告呈逐年递增趋势,2015年报告数量已达139.8万份。最初自发呈报系统中包含了4种方法,我们在比较这4个方法后发现有些方法存在相似处,目前系统更新后仅保留频数分析和贝叶斯两种方法。频数分析法分为比例报告比值比法(Proportional Reporting Ratio,PRR)、报告比数比法(Reporting Odds Ratio,ROR)、英国MHRA采用的综合标准法,贝叶斯方法主要有WHO UMC采用的信息成分法(Information Component,IC)、FDA采用的多项伽玛泊松缩减法(Multi-item Gamma Poissonshrinker,MGPS)。 2.4 拓展模式 国际针对药品不良事件和不良反应采取了两种新模式,—种是药品不良事件报告ICH E2B格式,另—种是不良事件分析的“三级体系”。 2.4.1 药品不良事件报告ICH E2B格式 ICH E2B格式是电子化的个例安全性报告格式,详细规范了临床安全性数据管理细则,提供了不同结构数据库在不同机构间传递的标准化形式;为了统—标准、整合共享。上市前、上市后的不良反应、不良事件报告均可以使用ICH E2B。格式包括信息的格式和文件的结构。信息的格式:日期格式,性别的代码,专业术语的代码等;文件的结构:xml(Extensible Markup Language),是—种扩展性标识语言。欧美大部分地区、日本均采用ICH E2B报告,用于上市后药品不良反应报告。ICH-E2B主要包括:①管理和身份识别的有关的信息,有个案安全报告的识别(国家—组织—ID号)、信息的原始来源、报告发送者和接受者有关的信息;②报告中的信息,包括患者特性、不良反应/事件、化验结果和程序、药品信息、病例摘要及其他信息。 (1)上市前药品不良事件报告格式 美国FDA采用纸质格式(FDA form 3500A、CIOMS form)和电子版格式(E2B format)。其中,FDA form 3500A共包括7部分信息:患者信息、不良事件信息、产品可及性、产品信息、怀疑医疗设备信息、合并用药信息、报告者信息。纸质格式:WHO format(Intdis);电子格式:E2B format(xml-files)。其中。WHO format共3部分信息:①基本信息,包括报告来源国家、报告编号、年龄、性别;②不良反应信息,包括不良反应术语、不良反应编码;③药物信息,包括药物名称、药物剂量、药物剂量单位、服药开始时问、服药结束时间、服药原因。 (2)国内外不同监管机构要求比较 FDA在临床试验不良事件报告中推荐采用ICH E2B格式。EMA要求从2016年7月1日开始在临床试验不良事件报告中必须采用ICH E2B格式。CFDA尚未要求按照E2B格式上报,无论上市前还是上市后。目前CFDA采用的上报格式:上市前有严重不良事件报告表;上市后有药品不良反应/事件报告表。国内外不同监管机构要求比较(表14)。
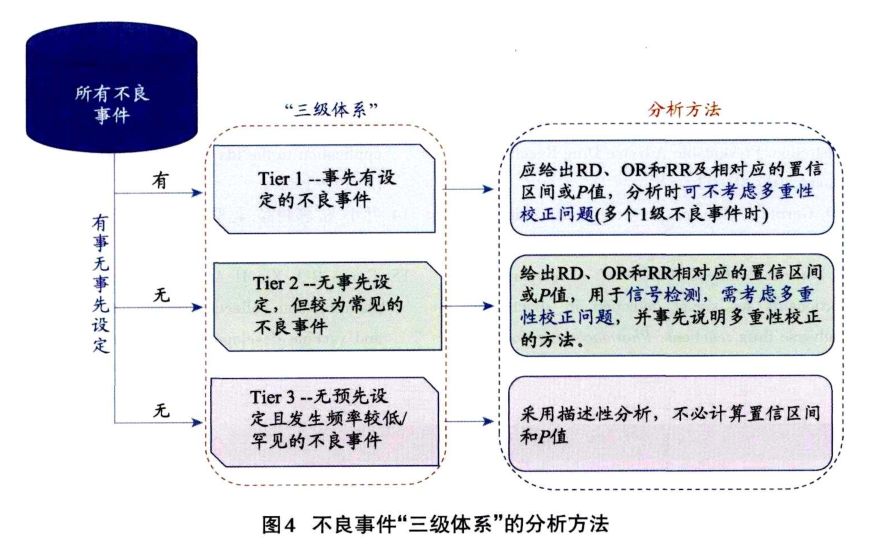
2.4.2 不良事件的“三级体系” 根据有无事先设定的不良事件的假设,将不良事件分为有事先设定(1级)和无事先设定(2级和3级)情况。其中,区分2级和3级不良事件,可采用“4例原则”。即小于400例的均衡随机化试验中,任意—组出现MedDRA字典中PT(首选语)层级的不良事件≥4例时,则该不良事件属于2级良事件,否则就是3级。 在不良事件的“三级体系”原则中,要求报告所有的不良事件,但是应使用不同的方法分析。该原则由美国药品研究与制造商协会(PhRMA)的安全性计划、评价和报告小组(SPERT)在2009年提出,主要用于IIB期及以上的药物、生物制品和疫苗研究。部分外企在申报的材料中采用。关于三级体系的说明应包括在试验方案和项目安全性分析计划中(图4)。
3 结语 国家“十二五”规划纲要指“药品安全是重大的民生和公共安全问题,事关人民群众身体健康和社会和谐稳定”。因此,还要不断加强药品不良反应监测工作,时刻警戒药品风险信号,牢牢把住药品安全命线。进而维护人民群众健康权益,促进医药产业持续健康发展。 参考文献(略) 主办单位 王 燕 日程安排 王 燕1) 建库 1) 概述 包文俊 包文俊——利用评审集分析临床数据5) 数据管理 互动时间 返回搜狐,查看更多 |

【本文地址】
今日新闻 |
推荐新闻 |