第一性原理 |
您所在的位置:网站首页 › 单个原子的结合能 › 第一性原理 |
第一性原理
|
基于第一性原理Sr2FeTaO6结合能和氧空位形成能的研究
概述计算重要参数结果分析总结与展望
概述
广义的第一原理包括两大类,以 Hartree-Fork 自洽场计算为基础的 ab initio 从头算和密度泛函理论(DFT)计算。也有人主张,ab initio 专指从头算,而第一性原理和所谓量子化学计算特指密度泛函理论计算。 第一性原理是指将多原子构成的体系理解为由电子和原子核组成的多粒子系统,在解体系薛定谔方程的过程中,最大限度地进行“非经验性”处理,即不涉及任何经验参数,所要输入的只是原子的核电荷数和一些模拟环境参量。 计算所求得的结果是体系薛定谔方程的本征值和本征函数 (波函数),有了这两项结果,就可研究体系的基本物理性质。 密度泛函理论(DFT)在许许多多的研究人员不断努力中得到了质的飞跃。随着现代科技的进步,尤其是芯片技术不断更新迭代使得计算机性能也得到了质的飞跃,并且人们对物理理论的认识也更加的深入。计算机模拟对材料进行设计是当代科学研究不可或缺的一种研究手段。相比较传统的实验方法,在许多情况下计算机模拟比实验更快、更省,并且还可以预测一些当前实验水平难以达到的理想情况。密度泛函的计算迭代过程如图一所示。 密度泛函在电池基础研究中有着广泛的应用,常用于计算电极材料的结构稳定性、嵌锂电位、电子结构、能带、弛豫结构、缺陷生成能、迁移路径、活化能以及锂离子传输动力学和脱嵌锂相变等性质。本文主要讲述使用VASP的计算流程,并简要分析结合能和氧空位。 计算重要参数在理论计算过程中,输入文件是必不可少的,其参数的设定也尤为重要。 2.1 计算过程中的主要参数 计算主要采用的是 VASP.5.4.4 程序,采用面波基组对SFT、SFM进行计算,为得到准确结果的同时又节约时间,需要对相关参数进行测试,需要测试的参数主要包括截断能、k 点以及泛函。赝势选择 VASP 自带的 PAW 势,每个原子的能量收敛标准为 0.0001 eV,每个原子的自洽收敛标准为 10-5 eV。由于体系中 Fe 元素为磁性元素,计算时应将自旋极化考虑在内。 使用VASP计算需要四个输入文件,INCAR文件控制了VASP进行何种性质的计算;POSCAR文件描述了所计算的体系的晶胞参数(包括基矢、晶格常数、原子类型、坐标位置);POTCAR文件包含了体系中各类元素的赝势;KPOINTS文件k点设置,可手动或自动产生。 2.1.1 截断能的选取 截断能的选取可以使得平面波动能小于设置的这个值,从而达到调节平面波基组的目的。Bloch 定理指出,每一个k 点的电子波函数可以展开成无限多形式的平面波基组,那也就意味着,无论截断能取多少,计算结果都会存在误差,增加截断能可以使计算结果更准确,由于体相中的原子个数较少,即使是较大的截断能,其计算量也比较小,因此在体相的计算中,截断能应适量地增加来提高计算精度。通过控制变量,分别对截断能为400eV、450eV、600eV进行实验,发现在优化时使用600eV的截断能比较合适,在计算氧空位、水和能等使用450eV的截断能即可。 2.1.2 K点的选取 在计算绝缘体的电子态时,选用很少的 k 点就能得到较为准确的结果,但是当计算体系为金属时,需要选取较为密集的 k 点才能获得准确的电子能带结构。在实际的计算中,k 点取值越多计算结果的精度越高,但同时也会带来计算量的增加。本次计算的K点是由vaspkit根据输入文件POSCAR自动生成的。 2.1.3 泛函的选取 自从20世纪60年代 (1964) 密度泛函理论 (DFT) 建立并在局城密度近似 (LDA) 下导出著名的Kohn - Sham (沈吕九)(KS)方程以来,DFT一直是凝聚态物理领域计算电子结构及其特性最有力的工具。它提供了第一性原理或从头算的计算框架。在这个框架下可以发展各式各样的能带计算方法。 局域密度近似 (LDA) 是所有的近似交换-关联泛函的基础。考虑一个电子密度缓变的系统,把写成局域量的积分形式,如公式。 局域 (自旋) 密度近似是在均匀电子气或电子密度变化足够缓慢的系统中提出的,用于描述密度变化大的非均匀电子气系统并不适合。因此在LDA的基础上,引入电子密度的梯度展开因子 ,提出了广义梯度密度近似 (GGA)。 由于交换相关能的两种近似方法 (LDA 和 GGA) 计算出来的晶格常数往往不同因此在进行计算时,需要对两种方法进行测试,其中 GGA 方法采用的是 PBE 泛函。经过分析,本文采用GGA-PBE泛函。 2.2 计算流程 首先构建模型,本文中的计算使用Materials Studio 8.0、VESTA进行模型搭建;然后准备四个输入文件,POSCAR使用VESTA生成,INCAR根据自己的需要修改相关参数,POTCAR和KPOINTS使用vaspkit辅助根据POSCAR中材料的结构自动生成,提交脚本vasp.pbs根据所用服务器的环境进行编写;最后使用提交命令提交任务。 结果分析3.1 结合能的概念 电池材料的结构稳定性是其能否应用的先决条件。内聚能是指自由原子组成化合物释放的能量,内聚能越高,化合物越稳定。形成能是单质形成化合物时释放的能量,形成能越高,化合物越稳定。相比于内聚能,用形成能判断材料的稳定性更符合实际,因为实际合成材料的时候,采用的是单质,而不是单个原子。内聚能和形成能是在绝对 0 度下的计算结果。对于简单的二元化合物AnBm(A,B为该化合物中包含的两种元素,n,m为相应原子在化学式中的数目)结合能公式如下: 多元化合物计算方法类似。 因为电池材料是放热反应,故结合能值为负值,以Sr2FeTaO6(SFT)、Sr2FeMoO6(STM)为例。经过计算,Sr2FeTaO6(SFT)、Sr2FeMoO6(STM)的相关数据如表一所示。 表一:物质能量 物质能量 能量/eV Sr -0.08780121 Fe -3.32207554 Mo -4.59412933 Ta -3.49488533 O2 -9.84275149 Sr4Fe2To2O12 -157.52469628 Sr4Fe2Mo2O12 -147.92116938 Sr18Fe10Ta10O60 -774.55006654 故经过计算Eb(Sr2FeTaO6)=-1.271327591eV;Eb(Sr2FeMoO6)=-0.791151246eV,Eb(Sr1.8FeTaO6)=-1.1656627210204eV,通过比较,Sr2FeTaO6的形成能较大,则其比较稳定。 3.2 氧空位 3.2.1 氧空位的概念 氧空位是物质中氧离子离开所留下的空位,一般认为带正电。氧空位指的是当晶格中的氧原子走后,顺手带走了两个原属于其他原子的电子,所留下的缺陷。换句话说,形成氧空位时,走掉的不只是氧,而是比氧多两个电子的氧离子O2-。 电催化剂中的氧空位(VO…)与析氢反应(HER)活性密切相关,然而,空位缺陷的作用及其浓度对催化剂析氢性能的影响仍不清楚。 氧空位(OV)是金属氧化物缺陷的一种,它们是金属氧化物在特定外界环境下(如高温、还原处理等)造成的晶格中氧的脱离,导致氧缺失形成氧空位。 缺陷方程可以表示为O=1/2O2+Vo.对于金属氧化物,其氧空位是缺陷的一种。 由于氧空位能够促进离子扩散和电子传输,最近在储能领域受到了广泛的关注。氧空位可以提高载流子的浓度,从而提高材料的电化学活性。通过应用密度泛函理论(DFT)可以计算电极材料的氧空位形成能。 可以根据公式确定一个氧空位的生成能形式 Edefect和Ehost分别表示失去一个氧原子和没有氧空位的超胞的能量。EO2表示一个气体氧分子的能量。 3.2.2 氧空位的计算 本次计算首先对Sr2FeTaO6(SFT)进行模型构建,然后进行扩胞,得到缺位后的材料Sr1.8FeTaO6(S1.8FT),计算使用的截断能为450eV。分别对O2、SFT、S1.8FT进行优化,得到体系总能量;然后在优化后的结构上去掉一个氧原子,再次进行优化,得到失氧后的体系能量。各部分能量如表二所示。 表二:各体系能量 Sr2FeTaO6 Sr1.8FeTaO6 O2 体系能量/eV -157.52469628 -774.55006654 -9.84275149 缺氧后能量/eV -148.73146838 -767.55540233 氧空位形成能 3.871852155 2.073288465 通过计算,从数据可以看出,Sr1.8FeTaO6的氧空位形成能较低,更容易形成氧空位,从而使电池性能有所提高。与上小节的结合能相互对应,Eb(Sr2FeTaO6)=-1.271327591eV、Eb(Sr1.8FeTaO6)=-1.1656627210204eV,可以发现Sr2FeTaO6的结合能更高一些,所以Sr2FeTaO6的结构相对于Sr2FeTaO6更稳定,更不易形成氧空位。 总结与展望4.1总结 DFT是当今处理相互作用多电子体系电子结构和几何结构最有力的工具。所谓从头算或第一性原理方法就是基于DFT框架建立起来的。它独立于实验,只需很少几个熟知的基本物理参数便可运作。 DFT并不要求原子的周期性排列,它具有十分广泛的适应性。已经在计算凝聚态物理、计算材料科学、量子化学、量子生物学和许多工业技术部门获得成功的应用。 4.2展望 随着各种理论的完善和计算机技术的发展,计算模拟已经成为电池研究中的重要方法之一。不同时间与空间尺度的模拟方法有助于我们从原子分子层次、介观层次和宏观层次理解材料结构与性能之间的关系。而这些不同尺度上的计算方法可以通过参数传递进行联系,从而实现材料的多尺度模拟,为材料设计提供了一种新的途径。通过实验与理论计算相结合的方法,有望解决的电池研究中的关键科学问题如下: (1)设计出新的电极与电解质材料:找到一些目前尚未发现的电池材料,并且具有更好的性能与更高的安全性。 (2)理解体相电极材料反应的热力学问题,通过精确计算材料的反应生成焓,预测材料在各种条件,如不同温度、不同压力、不同组分下的稳定性,甚至建议材料的合成方案。 (3)理解电位与材料组成、微观结构的关系。通过计算与实验得到大量已知结构及新结构所对应的电化学电位,建立电位与结构关系的数据库,采用统计方法,寻找决定电位的因素,理解它们之间的物理关联。 (4)电极材料充放电过程中组成与结构演变,通过计算得到的生成焓数据,建立与电极变化过程有关的相图,分析电极材料结构演变相对稳定性的影响,理解影响电极循环性能的因素以及电极材料的失效机制。 (5)与电池有关的复杂过程的理解,如输运机制,尺寸效应,界面问题等复杂过程。 (6)材料与器件制备过程、服役过程的数字化模拟与仿真。 |
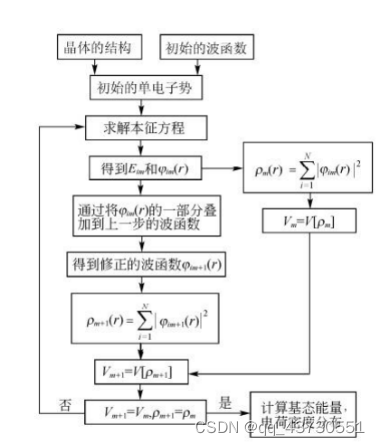 图1 密度泛函理论计算迭代过程示意图
图1 密度泛函理论计算迭代过程示意图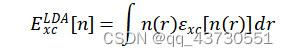
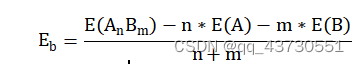
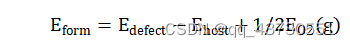
【本文地址】
今日新闻 |
推荐新闻 |