R语言 |
您所在的位置:网站首页 › 信阳至张家口 › R语言 |
R语言
|
提到热图,大家应该都不陌生。在文献中,热图常通过将数值映射为颜色,利用颜色变化直观地展示基因在不同样本中的的表达情况、基因与基因之间的相关性等等。那么,今天就给大家介绍一下如何使用pheatmap包来绘制热图。 1.安装并加载pheatmap包,并加载示例数据。 # 安装包 install.packages("pheatmap") # 加载包 library(pheatmap) # 加载数据 load("示例数据.rdata") is.matrix(data)绘制热图所需的数据结构是矩阵。这里的示例数据data就是一个行为基因,列为样本(T表示肿瘤样本,N表示正常样本)的10*10的矩阵(图1),中间的数值是每个基因在每个样本中表达的fpkm值。 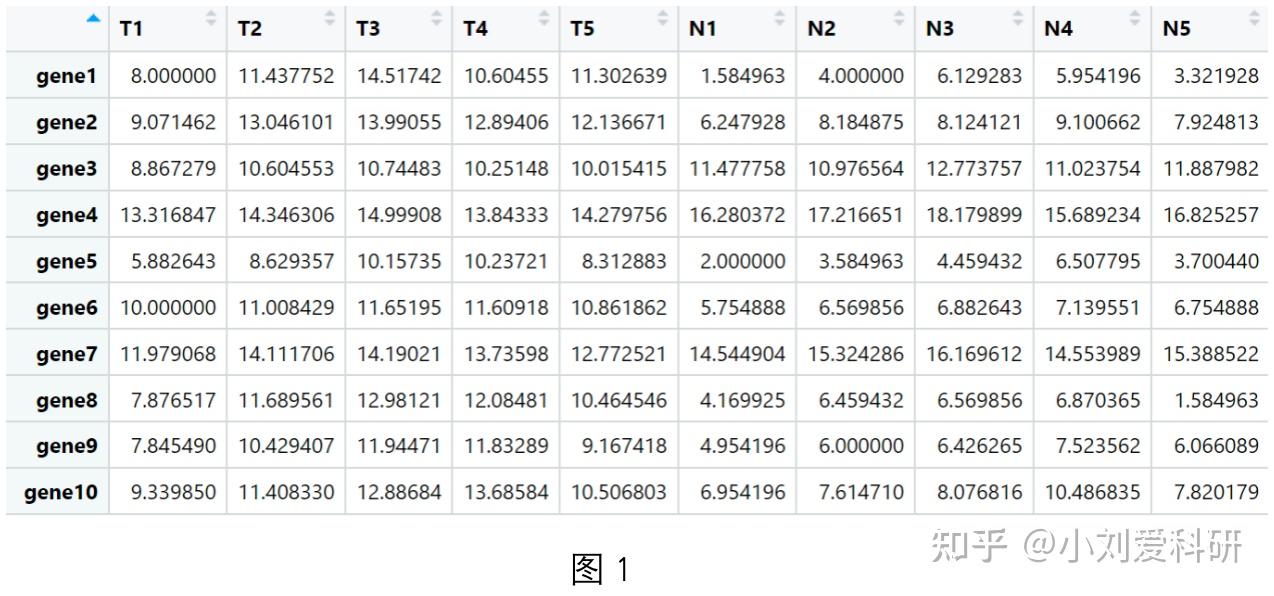 接下来,就可以使用pheatmap包中的pheatmap函数来画图了。我们先使用如下代码绘制一个相对简单的热图(图2)。 # 绘制热图 p1有几个问题需要注意一下: ① scale参数设置归一化方向。为什么要进行归一化呢? 我们前面介绍过,热图是利用颜色变化来直观地展示数值的变化。但如果绘制热图的数据差异过大,比如使用基因表达数据绘制热图,gene1在所有样本的表达量都在1000以上,gene2在所有样本的表达量都在1-10之间,由于数值之间相差过大,根本不在一个数量级上,那么在绘制热图时,两个基因在不同样本中表达的微小变化就很难通过颜色反映出来。所以,在绘制热图时,我们通常会对基因表达数据进行归一化,也就是将每一个基因表达量减去这个基因在所有样本中表达量的均值,然后除以其标准差,这种归一化的方式叫做标准正态化或Z-score处理。经过处理之后的数值都被等比例缩小了,每一个基因在所有样本的表达量就变成了均值为0,标准差为1的一组值。此时,绘制出来的热图就可以很好地展示所有基因在不同样本中表达的变化情况了。注意,如果某一个基因在所有样本中的表达量都相同,此时,由于标准差为0,不能作为分母,也就无法通过Z-score处理进行归一化,反映在热图上就是一整行或一整列灰色的色块。 ② 是按行还是按列归一化呢? 我们进行归一化的目的是将基因表达量拉到同一个数量级上。所以,行为基因,就按行进行归一化,列为基因,就按列进行。 ③ 关于聚类(cluster_rows 、cluster_cols参数)。 在上述代码中,我们将cluster_rows 、cluster_cols 的都设置成FALSE,也就是不对热图的行、列进行聚类。而在默认情况下,pheatmap包会通过基因表达数据计算各个样本以及各个基因之间的欧氏距离,然后根据欧式距离对样本和基因进行聚类(如图3)。可以发现,图3中基因和样本的顺序被重排,表达模式相似的基因、样本被排在一起,而表达模式差异大的基因,就会远离。在某些情况下,我们只是需要使用热图来直观呈现基因在样本中表达的变化规律,并不希望改变基因和样本的顺序,那就需要把参数cluster_rows 、cluster_cols 的设置成FALSE,不再进行聚类。 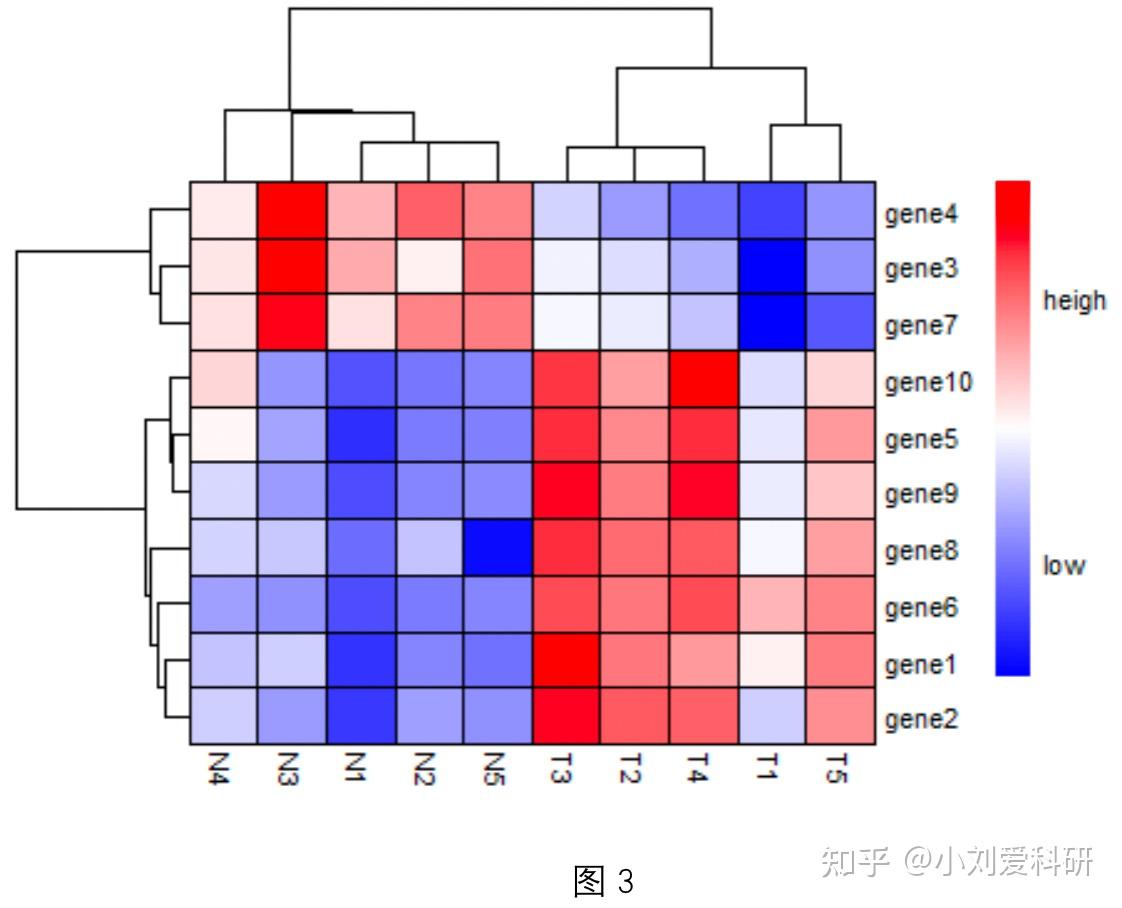 2.此外,我们还可以给热图添加注释信息。 首先,使用如下代码加载注释信息。 # 加载注释信息 load("列注释信息.rdata") load("行注释信息.rdata")如图4、5所示,分别是列注释信息anno_col与行注释信息anno_row,数据结构是数据框。 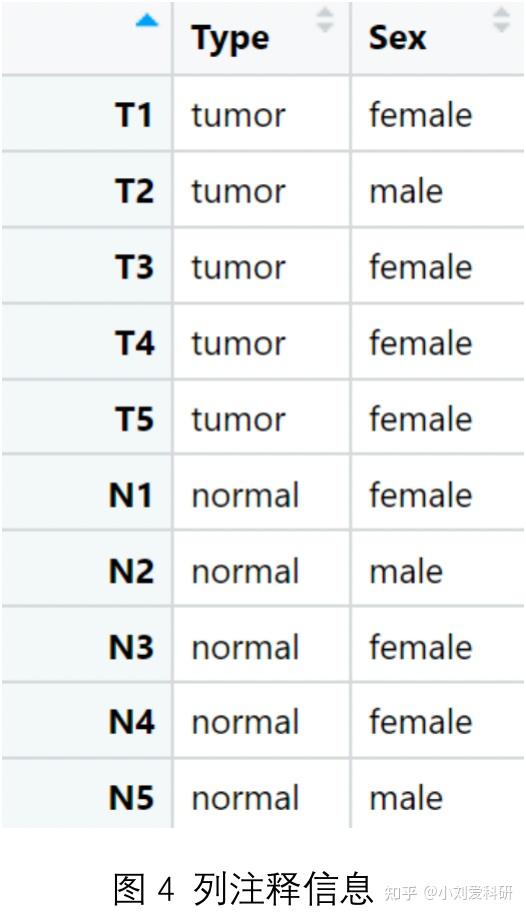  使用如下代码即可绘制出如图6所示的热图。 # 绘制热图 p23.最后,我们还可以在热图上展示每一个色块对应的数值。如图7,每个色块对应的经过归一化之后的数值都被标注在色块上了。 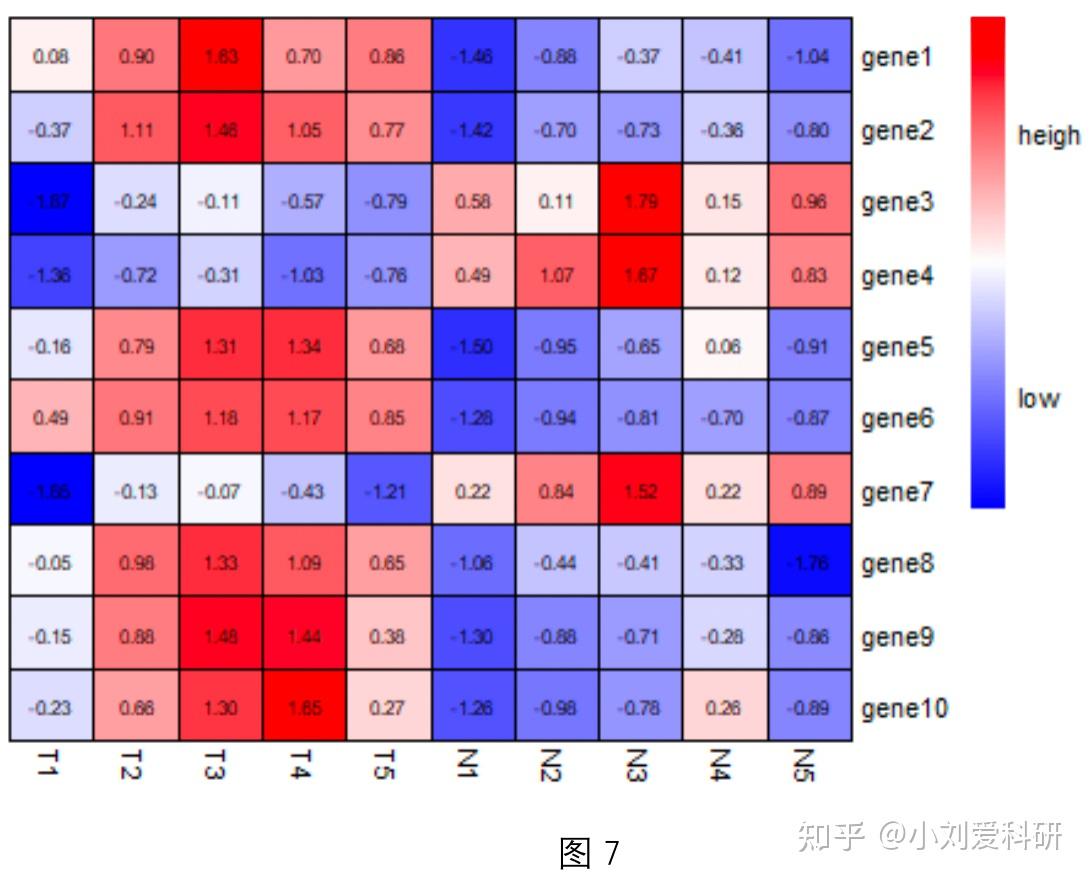 代码如下: # 绘制热图 p3然后再传递给display_numbers参数来实现(如图9)。 # 绘制热图 p4今天的分享到这里就结束了, 如果这期推文有帮助到你, 就请点一个免费的赞吧~ 我们下期再见! 本文首发于“生信大碗”公众号 转载请注明出处 |
【本文地址】
今日新闻 |
推荐新闻 |