浅析I期临床试验不良事件及风险管理 |
您所在的位置:网站首页 › 不良事件sac风险评估 › 浅析I期临床试验不良事件及风险管理 |
浅析I期临床试验不良事件及风险管理
|
3.排除标准 无明确不良反应/事件关系一览表的项目。 4.分析方法 对不良反应/事件进行分系统归纳,并分析其原因及风险要素构成,追溯不良事件的根源,探讨I期临床试验中风险管理的路径。所有数据采用百分比构成描述。 结果 筛选28个试验项目,剔除无不良反应/事件一览表的项目,共纳入24个试验项目590例健康受试者。通过整理试验药物的毒性成分、不良事件名称、所属系统、分级分度、例数、剂量情况、风险分类,分析7种因素与不良事件发生的关系。 1.受试者不良事件总体概况 纳人项目中,可分为3种试验:耐受性试验、药动学试验和生物等效性试验(本文统称为I期临床试验)。共发生不良事件39例。其中,耐受性试验有2种药物,共62例,受试者发生不良事件6例,不良事件发生率9.68%;药动学试验有16种药物,共352例,受试者发生不良事件2l例,不良事件发生率5.97%;生物等效性试验有6种药物,共176例,受试者发生不良事件12例,不良事件发生率为6.82%。 在发生的不良事件中,消化系统6例,占15.38%;皮肤黏膜6例,占15.38%;循环系统6例,占15.38%;神经系统2例,占5.13%;耳鼻喉2例(1例同时出现消化系统症状),占5.13%;全身症状1例,占2.57%;实验室检查异常值16例,占41.03%。 2.受试者不良事件发生的风险要素构成 不良事件发生的风险要素构成包括:①试验操作中的风险要素3例(其中1例与受试者自身相关)。②受试者自身存在的风险要素6例。③环境饮食因素5例。④试验设计中的风险要素4例。⑤试验用品本身存在的风险要素,如药物原因引起不良事件21例,可能药物引起者1例。 3.不同给药途径发生不良事件的情况 根据药物的给药途径分为3大类,即外用(皮肤敷帖给药、滴眼给药)、注射和口服。结果显示,不良反应发生率与给药途径相关:外用>口服>注射(见表1)。
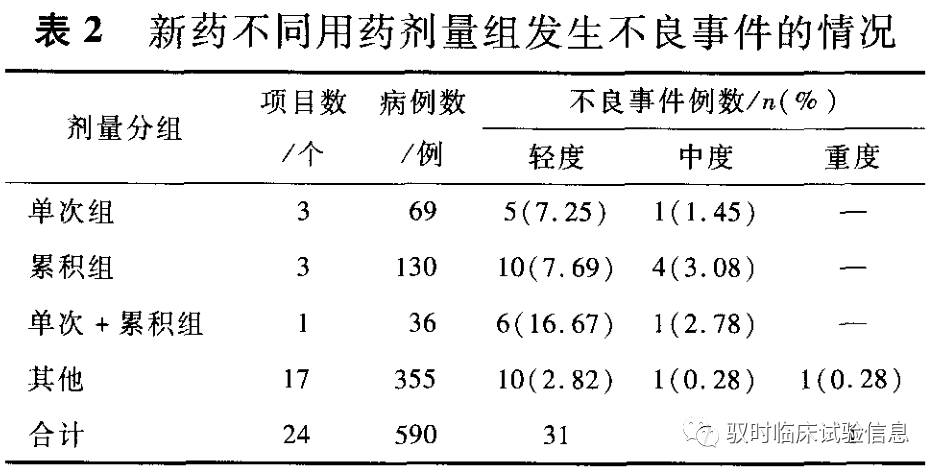
4.不同用药剂量组发生不良事件的情况 依据各试验项目总结报告中的用药方法,按“受试者疗程内服用药物剂量=新药每次用药量x每天用药次数x疗程(d)”公式,计算受试者疗程内服用药物剂量,分析不同用药剂量组不良事件的发生情况。见表2。
讨论 I 期临床试验是新药临床研究的起始阶段,存在许多潜在的、不可预知的风险。因此,不良反应/事件的监管及防范是一项重要课题。 1.受试者不良事件发生情况分析 我中心受试者不良反应/事件发生的概况有以下特点:①耐受性试验不良事件的发生率高于药动学试验和生物等效性试验,考虑与该试验药物首次应用于人体有关。并且,耐受性试验是人体试验的第一步,在该阶段被淘汰的药物不再进行后续研究。②不良事件发生率与给药途径相关,外用(13.33%)>口服(7.84%)>注射(4.15%)。外用药不良事件发生率最高,表现为皮肤瘙痒发红等局部症状,为药物与皮肤直接接触而致。注射剂不良事件发生率较低,与行业内对中药注射剂不良反应多见的认识不一致,分析可能与药物品种特点及注射剂使用方法相关。注射剂质量标准高和规范使用是减少不良事件的前提。③不良事件的发生率与给药剂量与疗程呈正相关。不同用药剂量组单次+累积组发生不良反应/事件率高于单次组、累积组与其他组,而累积组、单次+累积组发生不良事件的严重程度高,提示新药I期临床试验设计累积给药的必要性,给药时间间隔和疗程应结合临床拟使用疗程及药物半衰期来决定。以上不良事件的发生情况也可能与样本量小、研究者的主观偏移等因素有关。 2.受试者不良事件发生原因及风险要素构成情况分析 2.1试验操作中风险 研究护士操作规范与否直接关系到受试者的安危和试验药品的疗效。我中心受试者不良事件与操作相关者有3例。1例为女性受试者,由于血管较细,穿刺困难,夜班人员多次穿刺同侧上肢造成穿刺部位瘀肿,受试者疼痛难忍。另2例为男性受试者,1例在采集空白血时发生晕厥,随即苏醒;另1例发生休克,请急诊科会诊后诊断为重度腹泻致低血容量休克。该受试者有晕血史,试验前1d多次重度腹泻,未进食,但未告知研究人员。因此,受试者不良事件的发生也与受试者主观因素有关。如果研究必须要纳入有晕血史的受试者,应避免立位或坐位采血,应采取卧位并且选择方便实施救治的场所进行。研究过程中,操作者对受试者的观察、询问须到位。 2.2受试者自身风险 临床试验不良事件的发生与志愿者自身因素相关,如受试者年龄、性别、受教育程度、经济状况、种族、性格、有无不良嗜好等。I期临床试验受试者为健康志愿者,他们参与试验时最大的担心就是试验药物是否会对自己的身体造成损害,同时频繁抽血让其产生焦虑、恐惧心理。6例受试者中有2例试验期间出现“不适”,自觉无力或心悸,经研究者判断与药物无相关性。另有3例因其他受试者出现腹胀、腹泻而出现“腹胀不适”表现。1例受试者在试验洗脱期打篮球时不慎致眉棱骨受伤合并用药而退出研究。 2.3环境饮食因素 不良事件发生的环境比较复杂,除药物的影响外,还存在着其他可能的影响因素,如饮食、合用药物、患者罹患的疾病和并发症等。在新药I期临床试验中,病房环境及饮食因素亦是影响不良事件发生的重要因素。5例不良事件表现为口干、腹泻、腹部不适和稀便。追问不良事件的发生情况,口干与冬季空调环境下空气湿度小、受试者不适应环境变化有关;表现为消化系统症状者有3个原因:夏季开空调未盖被子、夜问开窗休息与进食牛奶。当然,不良事件的发生原因与受试者自身身体状态、病房管理等因素亦有相关性,是一个单因素或多因素影响的事件。 2.4试验设计中的风险 药物临床试验方案是指导临床试验实施的纲领性文件,试验方案的好坏是药物临床试验能否取得成功的决定性因素。方案设计质量和受试者的不良事件/反应发生亦密切相关。如受试者风险与试验期间受试者的管理密切相关。如果方案设计中明确规定受试者管理的操作细则,可在一定程度上避免不良事件的发生;在满足试验要求的条件下,方案应尽量详细写明受试者的餐谱、进餐时间和进餐量,可以减少受试者的饮食因素可能导致的肝酶轻度升高等不良事件的出现。科学严谨的方案设计与实施是试验质量和受试者安全的重要保障。 2.5药品自身风险 药品作为一种特殊商品,本身即具有一定的风险属性。新药未经过大范围的临床使用,对其不良反应的了解也不够深入。因此,药品本身的风险成为常见的不良事件风险要素。我中心受试者不良事件由药物引起者占总数的56.41%(包括1例可能药物引起者)。在耐受性试验中,不良反应主要表现为口干、咽痛、头痛、耳鸣、皮肤瘙痒等,考虑与试验药物含有蟾酥内酯、马钱子、乌头碱成分相关,与文献报道一致。药动学试验、生物等效试验出现的药物相关性不良反应主要表现为胸闷、心悸、口干、心室内传导阻滞、T波倒置、sT—T改变、QT间期延长、手发抖、活动欠灵活、腹泻、全身乏力、血清ALT和AST增高、尿蛋白含量增高。 3.风险管理 汶柯等探讨了在我国新药临床试验中进行受试者风险最小化管理的方法,发现临床试验的高风险及不确定性使受试者常暴露于高风险中。因此,关于风险管理措施的制定及实施监管成为减少不良事件的重要举措。其中最为重要的是临床试验方案的制定及试验流程的过程管理。本研究针对所述风险要素探讨相应的管理措施。 3.1试验操作中的风险 操作过程的管理与控制是保证试验质量和减少不良事件发生的关键所在。因此,所有试验过程都要进行严格的过程监管。 试验前详细的授权分工和分工确认培训会是减少实施过程中风险的必要前提。试验分工步骤要详尽并且责任到人,培训会要让所有参加试验的工作人员了解药物研究背景知识及注意事项,提高防范意识。操作人员过硬的专业技术水平是减少不良事件发生的重要保证。因此,应进行内部练兵,对操作人员定期培训考核、绩效分配结合工作实效,对责任失误加以严惩,以提高操作水平。规范操作依赖于严格到位的操作SOP,因此相关SOP的制定和落实是减少操作风险的基石。完善的制度管理也很重要,低年资护士夜班时,相应安排二线值班护士,以应对临时突发情况,最大限度减少不良事件的发生,这样遇到穿刺困难、受试者不配合等情况时就能沉着应对。试验前对受试者问询、查看也是一个非常重要的环节。研究者(包括研究护士)在试验前要仔细观察每一位受试者的状态,并与之交谈,以发现近期可能发生的一些异常状态,敏于思考、汇总,必要时会同上级医师判断,防患于未然。 3.2受试者自身存在的风险 I 期临床试验的试验药物通常是创新药,是首次在人体使用,因而受试者风险相对较大。而受试者本身亦存在潜在的风险隐患因素。研究者在与受试者沟通时要详细告知可能发生的不良反应及注意事项,尽量规避一些潜在风险,做到风险最小化,同时注意排除具有潜在风险的受试者。研究者可采取以下措施:①筛选受试者时,全面了解受试者的一般情况,主要是个人和环境因素,尽量选择依从性佳的受试者。②采取多种方式进行依从性教育,使受试者及其家属了解试验的意义,嘱其一旦出现任何不适,及时与研究者联系。③让受试者充分知情并对其进行培训:尽可能地详尽告知试验药物本身的药理毒理作用、前期试验结果、本次试验内容、用药方案、参与试验的受益与风险,从而减少试验潜在风险。 3.3环境饮食因素 良好的环境和适宜的饮食是受试者试验完成的重要保证。因此试验期间的管理工作应该到位,包括受试者管理、病房环境管理、餐食饮水管理,都应按照方案要求、制定相应的计划,做到事无巨细,防患风险于未然。试验期间应时时考虑到环境、饮食因素引起受试者不良事件的可能性,如冬季病房环境温度较低时,可以利用电暖气、暖手袋等为受试者取暖,保证血样采集顺利,减少受试者不适和痛苦。 试验方案制定时应对受试者食谱做出规定,除高脂高热饮食要求外,应统一清淡饮食,并对受试者的餐食来源的可靠性予以要求。不同类型试验的进餐时间、服药时间间隔、进食数量与种类都应保证受试者能量来源且避免受试者超量进餐、选择进餐或暴饮暴食,以规避由于饮食因素引发的不良事件。 3.4试验设计中的风险要素 临床试验方案是临床试验的重要文件,试验中进行的程序、测试步骤、资料数据的收集等均要按照方案要求操作。开始撰写试验方案前,要联系申办方获得药物相关资料,如临床前药理毒理、药学资料、既往临床研究、同品种研究情况等基本信息。结合试验目的、安全性等确定合适的剂量(包括剂量递增计划)、疗程、洗脱期及生物标本采集时点及量。同时在方案设计之初,邀请临床试验专家、申办方、试验医院研究者等各方介入,对试验方案进行反复研讨和确认,增加试验方案的科学依据。 本研究结果显示,单次+累积给药组不良事件的发生率为19.45%,高剂量组不良事件的发生率为10.77%,提示研究方案设计要紧密结合药物临床前毒理、药动学研究结果,考虑药物半衰期和安全剂量范围;在达到研究目的的同时,尽量减少给药剂量、次数及给药时间,可在一定程度上降低不良事件的发生。方案中要重点关注预期的和非预期的不良反应,体现监测计划、应急预案及救治措施,客观制定符合实际的临床试验中止标准。 3.5药品本身风险 药品本身不良反应难以避免,但科学合理的试验设计可在一定程度减少发生不良反应的受试者例数,如耐受性试验中合理的起始剂量估算、剂量递增梯度的把握、对药品临床前毒理的考虑等;药动学试验在符合法规要求和满足检测需要的基础上,对给药组剂量的选择等。药品本身风险和使用过程亦相关,合理的试验设计和规范操作可在一定程度上减少不良事件的发生。 综上,保护受试者权益,以受试者的健康为本,不仅符合《赫尔辛基宣言一人体医学研究的伦理准则》的要求,也符合现代医学发展的趋势。我们应该在试验早期即重视受试者的安全,针对不良事件发生可能的风险要素构成,认真落实试验进展的每个阶段和细节,降低可能存在的风险。机构和伦理委员会也应加强对试验风险和受试者保护方面的审查。笔者认为,风险管理要贯穿整个临床试验,从前期的试验方案设计到试验过程及后期的试验结题,药物临床试验中引入风险管理理论可有效减少风险的发生,最大化地降低损失。为此,我们有必要建立风险的预防、评估与管理机制,建立科学的风险决策机制,强化管理,提高医疗技术临床试验的风险评估与安全管理的有效性,推动我国临床试验的发展。 参考文献(略) 接受日期:2016-11-16 精品干货分享
详情介绍和购买返回搜狐,查看更多 |



【本文地址】