2,3,6,7 |
您所在的位置:网站首页 › 丁炔二酸二乙酯cas › 2,3,6,7 |
2,3,6,7
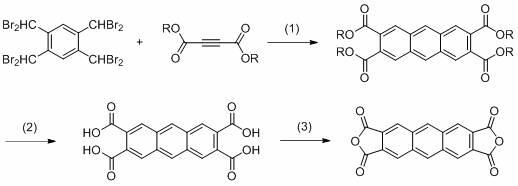 1.本发明属于有机合成技术领域,具体涉及一种2,3,6,7-蒽四羧酸二酐的合成方法。 背景技术: 2.高性能有机高分子材料具有优异的力学性能、耐热性能、耐辐射性能、及耐水解性能等,在塑料,薄膜,泡沫,纤维,复合材料,液晶和光刻胶等领域得到了广泛的应用,其中,有机高分子发光材料具有工作电压低,能耗低,良机械加工性等特性,已被广泛研究并应用于各类显示器件中。3.蒽作为典型蓝光生色基团,具有较宽的能隙、优良的热稳定性和较高的发光效率,一直备受人们重视。目前,含蒽类的有机发光材料实用化并不高,这主要是因为目前开发的含蒽类发光材料发光效率和使用寿命都比较低。因此,继续开发稳定、高效的蒽类发光材料具有十分重要的意义。4.2,3,6,7-蒽四羧酸二酐,简称antda,cas号为4430-56-2,分子式为c18h6o6,分子量为318.24,黄色固体,熔点>300℃,结构式如下:。5.作为一种新型的蓝色发光聚酰亚胺二酐单体,2,3,6,7-蒽四羧酸二酐具有较好的应用前景。现有技术公开的2,3,6,7-蒽四羧酸二酐的合成方法如下:日本专利文献jp2008297354a公开了一种2,3,6,7-蒽四羧酸二酐的合成方法,它是以1,2,4,5-四甲基苯为起始原料,先经溴化反应得到1,2,4,5-四(二溴甲基)苯,然后与n-苯基马来酰亚胺在碘化钠的催化下,经狄尔斯-阿尔德(diels-alder)反应得到n,n'-二苯基-2,3,6,7-蒽二酰亚胺,接着水解得到 2,3,6,7-蒽四羧酸,最后脱水成酐得到2,3,6,7-蒽四羧酸二酐。6.该方法的不足在于:(1)制备n,n'-二苯基-2,3,6,7-蒽二酰亚胺的收率较低,只有35%;(2)n,n'-二苯基-2,3,6,7-蒽二酰亚胺的溶解度极差,不利于中控和纯化;(3)n,n'-二苯基-2,3,6,7-蒽二酰亚胺的水解反应条件也较为苛刻,需要在150℃高温下反应25h,对生产设备要求较高,而且水解产生的废水主要成分为苯胺,毒性较大,对环境和人体不友好。 技术实现要素: 7.本发明的目的在于解决上述问题,提供一种收率较高、水解反应条件较为温和、对人体和环境较为友好的2,3,6,7-蒽四羧酸二酐的合成方法。8.实现本发明目的的技术方案是:一种2,3,6,7-蒽四羧酸二酐的合成方法,具有以下步骤:①以1,2,4,5-四(二溴甲基)苯为起始原料,先与丁炔二羧酸酯进行狄尔斯-阿尔德反应,得到2,3,6,7-蒽四羧酸四酯;②步骤①得到的2,3,6,7-蒽四羧酸四酯水解,得到2,3,6,7-蒽四羧酸;③步骤②得到的2,3,6,7-蒽四羧酸脱水成酐,得到2,3,6,7-蒽四羧酸二酐。9.合成路线如下:。10.其中:r表示甲基【-ch3】、乙基【-ch2ch3】或者叔丁基【-c(ch3)3)】。11.上述步骤①中,所述1,2,4,5-四(二溴甲基)苯与所述丁炔二羧酸酯的摩尔比为1∶2~1∶5;所述丁炔二羧酸酯为丁炔二酸二甲酯或者丁炔二酸二乙酯或者丁炔二酸二叔丁酯。12.上述步骤①中所述狄尔斯-阿尔德反应是在碘化钠的存在下进行的;所述1,2,4,5-四(二溴甲基)苯与所述碘化钠的摩尔比为1∶8~1∶12。13.上述步骤①中所述狄尔斯-阿尔德反应是在有机溶剂中进行的;所述有机溶剂为dmf或者dmac。14.上述步骤①中所述狄尔斯-阿尔德反应的温度为60~120℃。15.上述步骤②中所述水解的反应温度为室温~80℃,优选为室温~55℃,更优选为室温。16.上述步骤②中所述水解为碱性水解或者酸性水解。17.所述碱性水解是在无机碱的存在下进行的;所述无机碱为氢氧化锂、氢氧化钠或者氢氧化钾;所述2,3,6,7-蒽四羧酸四酯与所述无机碱的摩尔比为1∶3~1∶10。18.所述碱性水解是在混合溶剂中进行的;所述混合溶剂由甲醇、乙醇或者四氢呋喃中的一种与水组成。19.所述酸性水解是在无机酸的存在下进行的;所述无机酸为盐酸或者醋酸;所述2,3,6,7-蒽四羧酸四酯与所述无机酸的摩尔比为1∶3~1∶10。20.所述酸性水解是在有机溶剂中进行的;所述有机溶剂为甲醇、乙醇或者四氢呋喃。21.上述步骤③中所述脱水成酐采用本领域常规方法。22.本发明具有的积极效果:(1)本发明的合成路线采用丁炔二羧酸酯作为亲双烯体,得到的中间体2,3,6,7-蒽四羧酸四酯相比于现有技术的n,n'-二苯基-2,3,6,7-蒽二酰亚胺,溶解度和极性都适中,利于反应中控和分离纯化,适合工业化大生产,而且该反应收率相对较高,可达50%左右。23.(2)本发明的中间体2,3,6,7-蒽四羧酸四酯水解反应条件较为温和(室温即可),而且反应时间大大缩短,尤其是水解反应的废液主要成分为醇类,毒性明显低于现有技术的苯胺,对环境和人体更为友好。附图说明24.图1为实施例1步骤①制得的2,3,6,7-蒽四羧酸四甲酯的lc-ms谱图。25.图2为实施例1步骤②制得的2,3,6,7-蒽四羧酸的lc-ms谱图。26.图3为实施例1步骤③制得的目标产物2,3,6,7-蒽四羧酸二酐使用甲醇配样后的lc-ms谱图。27.图4为实施例2步骤①制得的2,3,6,7-蒽四羧酸四乙酯的lc-ms谱图。28.图5为实施例3步骤①制得的2,3,6,7-蒽四羧酸四叔丁酯的lc-ms谱图。具体实施方式29.(实施例1)本实施例的2,3,6,7-蒽四羧酸二酐的合成路线如下:。30.具体合成方法如下:①氮气保护下,将80.0g的1,2,4,5-四(二溴甲基)苯(0.104mol)、29.7g丁炔二酸二甲酯(0.209mol)、157g碘化钠(1.05mol)和800ml的dmac加入到2l三口瓶,80℃搅拌反应10h。31.反应结束后,向反应体系中加入500g水和500g乙酸乙酯,搅拌30min静置,分去下层水相,将有机相经减压蒸馏后,加入二氯甲烷和甲醇混合液重结晶,得到金黄色固体21.9g,纯度为99.8%(hplc),收率为51.3%,熔点为139~141℃,lc-ms谱图见图1。32.由图1可以看出:该产物的分子量为411.2,与2,3,6,7-蒽四羧酸四甲酯相吻合。33.②将20.0g步骤①制得的2,3,6,7-蒽四羧酸四甲酯(0.049mol)加入到250ml三口瓶中,然后加入150g甲醇、100g水以及9.75g氢氧化钠(0.244mol),氮气保护下,室温搅拌反应2~3h,溶液变为澄清。34.反应结束后,40℃下减压蒸馏除去甲醇,将36wt%浓盐酸滴入水相中直至ph=3~4,有黄色固体析出,过滤,滤饼用80g水漂洗,70℃真空干燥15h,得到黄色固体17.0g,纯度为99.5%(hplc),收率为98.4%,熔点为242~244℃,lc-ms谱图见图2。35.由图2可以看出:该产物的分子量为355.0,与2,3,6,7-蒽四羧酸相吻合。36.③将17.0g步骤②制得的2,3,6,7-蒽四羧酸(0.048mol)和136g乙酸酐(1.33mol)加入到250ml三口瓶中,升温至120±2℃搅拌反应3h。37.反应结束后,降至室温,过滤,滤饼用30g甲苯漂洗,60℃真空干燥15h,得到12.6g黄色粉末状固体2,3,6,7-蒽四羧酸二酐,纯度为99.5%(hplc),收率为82.5%。38.对该目标产物使用甲醇配样后进行lc-ms检测,结果见图3。39.由图3可以看出:分子量为383.1,与2,3,6,7-蒽四羧酸二酐使用甲醇配样相吻合。[0040]1h nmr:(dmso-d6)9.09(s,2h),9.42(s,4h)ppm。[0041](实施例2)本实施例的2,3,6,7-蒽四羧酸二酐的合成路线如下:。[0042]具体合成方法如下:①氮气保护下,将80.0g的1,2,4,5-四(二溴甲基)苯(0.104mol)、35.6g丁炔二酸二乙酯(0.209mol)、157g碘化钠(1.05mol)和800ml的dmf加入到2l三口瓶,80℃搅拌反应10h。[0043]反应结束后,向反应体系中加入500g水和500g乙酸乙酯,搅拌30min静置,分去下层水相,将有机相经减压蒸馏后,加入二氯甲烷和甲醇混合液重结晶,得到金黄色固体24.1g,纯度为99.8%(hplc),收率为49.5%,熔点为154~156℃,lc-ms谱图见图4。[0044]由图4可以看出:该产物的分子量为467.2,与2,3,6,7-蒽四羧酸四乙酯相吻合。[0045]②将24.0g步骤①制得的2,3,6,7-蒽四羧酸四乙酯(0.052mol)加入到250ml三口瓶中,然后加入50g甲醇、100g水以及10.3g氢氧化钠(0.258mol),氮气保护下,室温搅拌反应2~3h,溶液变为澄清。[0046]反应结束后,40℃下减压蒸馏除去甲醇,将36wt%浓盐酸滴入水相中直至ph=3~4,有黄色固体析出,过滤,滤饼用60g水漂洗,70℃真空干燥15h,得到黄色固体18.1g,纯度为99.7%(hplc),收率为99.3%,熔点为242~244℃。[0047]③将18.0g步骤②制得的2,3,6,7-蒽四羧酸(0.051mol)和144g乙酸酐(1.41mol)加入到250ml三口瓶中,升温至120±2℃搅拌反应3h。[0048]反应结束后,降至室温,过滤,滤饼用30g甲苯漂洗,60℃真空干燥15h,得到14.8g黄色粉末状固体2,3,6,7-蒽四羧酸二酐,纯度为99.7%(hplc),收率为91.5%。[0049](实施例3)本实施例的2,3,6,7-蒽四羧酸二酐的合成路线如下:。[0050]具体合成方法如下:①氮气保护下,将80.0g的1,2,4,5-四(二溴甲基)苯(0.104mol)、47.3g丁炔二酸二叔丁酯(0.209mol)、157g碘化钠(1.05mol)和800ml 的dmf加入到2l三口瓶,80℃搅拌反应10h。[0051]反应结束后,向反应体系中加入500g水和500g乙酸乙酯,搅拌30min静置,分去下层水相,将有机相经减压蒸馏后,加入二氯甲烷和甲醇混合液重结晶,得到金黄色固体28.4g,纯度为99.2%(hplc),收率为47.0%,lc-ms谱图见图5。[0052]由图5可以看出:该产物的分子量为579.3,与2,3,6,7-蒽四羧酸四叔丁酯相吻合。[0053]②将28.0g步骤①制得的2,3,6,7-蒽四羧酸四叔丁酯(0.048mol)加入到250ml三口瓶中,然后加入280g甲醇,氮气保护下,加入盐酸甲醇溶液(2m,120ml),室温搅拌反应6h。[0054]反应结束后,过滤,滤饼用60ml甲醇漂洗,70℃真空干燥15h,得到黄色固体17.0g,纯度为99.7%(hplc),收率为99.1%,熔点为242~244℃。[0055]③将17.0g步骤②制得的2,3,6,7-蒽四羧酸(0.048mol)和136g乙酸酐(1.33mol)加入到250ml三口瓶中,升温至120±2℃搅拌反应3h。[0056]反应结束后,降至室温,过滤,滤饼用30g甲苯漂洗,60℃真空干燥15h,得到12.5g黄色粉末状固体2,3,6,7-蒽四羧酸二酐,纯度为99.5%(hplc),收率为81.9%。 |
【本文地址】
今日新闻 |
推荐新闻 |