InferCNV 的 Gene ordering file 输入文件制作 |
您所在的位置:网站首页 › vtt文件制作 › InferCNV 的 Gene ordering file 输入文件制作 |
InferCNV 的 Gene ordering file 输入文件制作
|
我们都知道,利用R包infercnv对scRNA-seq数据进行CNV推断时,首个步骤是运行CreateInfercnvObject()函数构建infercnv对象,此处必须设置gene_order_file参数,其输入是一个基因的染色体位置信息文件,以制表符分隔。 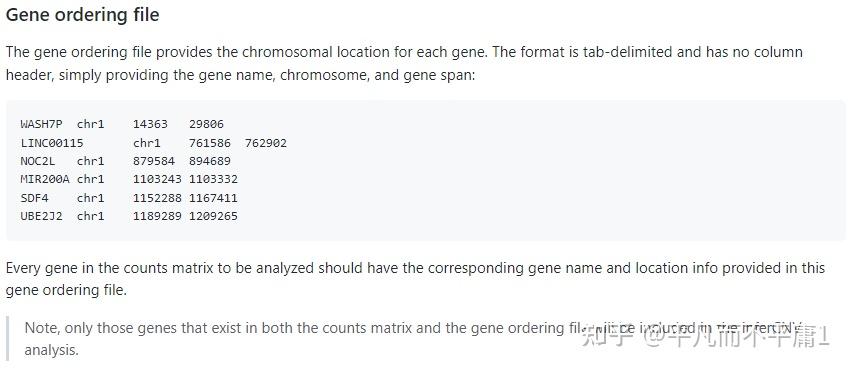 inferCNV作为TrinityCTAT Toolkit的一个组成部分,一些版本的 Genomic Position Files 已经生成过并且放置在 https://data.broadinstitute.org/Trinity/CTAT/cnv/ 供大家获取。  可以看出数据的版本比较老,有些基因组注释文件还是依赖hg19参考基因组,而我们现在表达定量,特别是10x数据,上游一般直接用Cell Ranger流程,官网目前给出的集成好的参考基因组相关内容的压缩包refdata-gex-GRCh38-2020-A.tar.gz内的文件都是基于hg38。如果还用TrinityCTAT给出的老数据作为InferCNV gene_order_file参数的输入,得到的结果总会是相对粗糙的。那如果我们想自己构建一个 Genomic Position File 呢?贴心的 developer 造了一个现成的轮子,我们把轮子下载下来。同时下载与10x官网给出信息一致对应的gencode.v32文件。 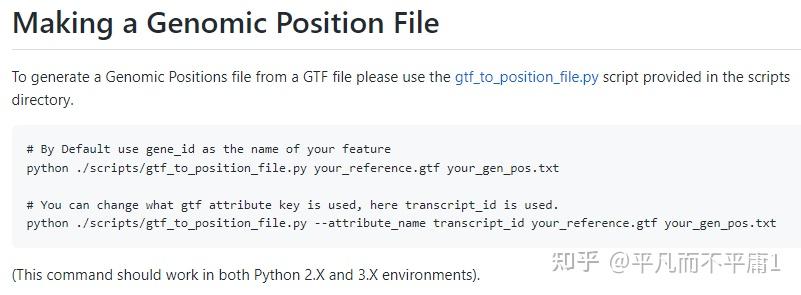 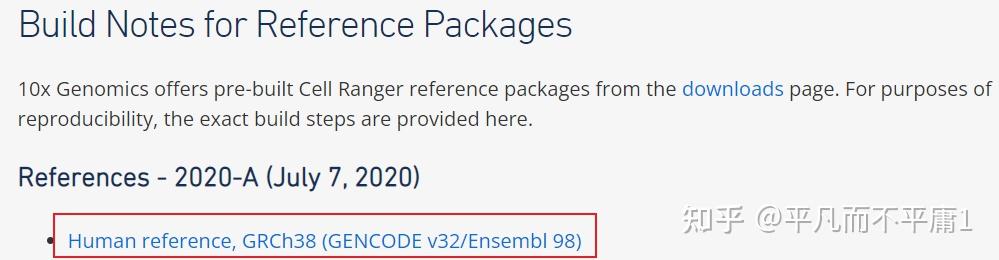 cd /mnt/d/Bioinfo/Single_Cell/inferCNV/
wget -c https://github.com/broadinstitute/infercnv/raw/master/scripts/gtf_to_position_file.py
wget -c http://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_32/gencode.v32.primary_assembly.annotation.gtf.gz cd /mnt/d/Bioinfo/Single_Cell/inferCNV/
wget -c https://github.com/broadinstitute/infercnv/raw/master/scripts/gtf_to_position_file.py
wget -c http://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_32/gencode.v32.primary_assembly.annotation.gtf.gz
运行时报错,443端口http://raw.githubusercontent.com域名解析有问题。 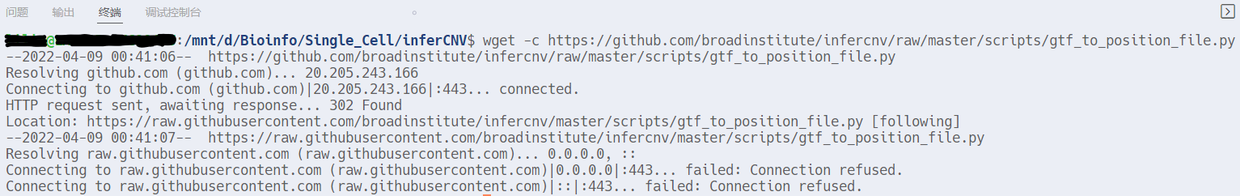 因为通常情况下我们会直接用git clone下载整个repository,很少下载单个文件,所以第一次遇到这个报错。需要以sudo权限打开/etc/hosts文件,sudo vim /etc/hosts,然后在末尾添加一行199.232.68.133 raw.githubusercontent.com。再次下载,成功。 看下脚本的用法。运行python脚本,生成我们自己DIY的Genomic Position File。默认使用gtf文件的gene_id字段,也可通过--attribute_name设置其它,例如gene_name字段。  python gtf_to_position_file.py -h
# By Default, gene_id
python gtf_to_position_file.py gencode.v32.primary_assembly.annotation.gtf gencode_v32_gene_pos_gene_id.txt
# gene_name
python gtf_to_position_file.py --attribute_name gene_name gencode.v32.primary_assembly.annotation.gtf gencode_v32_gene_pos_gene_name.txt python gtf_to_position_file.py -h
# By Default, gene_id
python gtf_to_position_file.py gencode.v32.primary_assembly.annotation.gtf gencode_v32_gene_pos_gene_id.txt
# gene_name
python gtf_to_position_file.py --attribute_name gene_name gencode.v32.primary_assembly.annotation.gtf gencode_v32_gene_pos_gene_name.txt
得到生成的文件如下,可以进行之后的infercnv步骤。  把源代码贴在下面学习一下,脚本相对比较简单,只调用了常规的argparse,csv,os三个标准库。写成一个函数convert_to_positional_file()解决问题。 #!/usr/bin/env python """ Converts GTF files to proprietary formats. """ # Import statements import argparse import csv import os __author__ = 'Timothy Tickle, Itay Tirosh, Brian Haas' __copyright__ = 'Copyright 2016' __credits__ = ["Timothy Tickle"] __license__ = 'BSD-3' __maintainer__ = 'Timothy Tickle' __email__ = '[email protected]' __status__ = 'Development' def convert_to_positional_file(input_gtf, output_positional, attribute_key): """ Convert input GTF file to positional file. :param input_gtf: Path to input gtf file :type input_gtf: String :param output_positional: Path to output positional file :type output_positional: String :param attribute_key: Key of the GTF attribute to use for feature/row names :type attribute_key: String :returns: Indicator of success (True) or Failure (False) :rtype: boolean """ if not input_gtf or not os.path.exists(input_gtf): print("".join(["gtf_to_position_file.py:: ", "Could not find input file : " + input_gtf])) all_genes_found = set() # Holds lines to output after parsing. output_line = [] previous_gene = None previous_chr = None gene_positions = [] # Metrics for the file i_comments = 0 i_duplicate_entries = 0 i_entries = 0 i_accepted_entries = 0 i_written_lines = 0 with open(input_gtf, "r") as gtf: gtf_file = csv.reader(gtf,delimiter="\t") for gtf_line in gtf_file: if gtf_line[0][0] == "#": i_comments += 1 continue i_entries += 1 # Clean up the attribute keys and match the one of interest. attributes = gtf_line[8].split(";") attributes = [entry.strip(" ") for entry in attributes] attributes = [entry.split(" ") for entry in attributes if entry] attributes = [[entry[0].strip('"'),entry[1].strip('"')] for entry in attributes] attributes = dict([[entry[0].split("|")[0],entry[1]] for entry in attributes]) if attribute_key in attributes: gene_name = attributes[attribute_key] else: print("Could not find an attribute in the GTF with the name '"+attribute_key+"'. Line="+"\t".join(gtf_line)) exit(99) if not gene_name == previous_gene: if len(gene_positions) > 1 and previous_gene not in all_genes_found: i_accepted_entries += 1 gene_positions.sort() output_line.append("\t".join([previous_gene, previous_chr, str(gene_positions[0]), str(gene_positions[-1])])) all_genes_found.add(previous_gene) gene_positions = [] else: i_duplicate_entries += 1 gene_positions += [int(gtf_line[3]), int(gtf_line[4])] previous_gene = gene_name previous_chr = gtf_line[0] if previous_gene and previous_chr and len(gene_positions) > 1: i_accepted_entries += 1 gene_positions.sort() output_line.append("\t".join([previous_gene, previous_chr, str(gene_positions[0]), str(gene_positions[-1])])) with open(output_positional, "w") as positional_file: i_written_lines += len(output_line) positional_file.write("\n".join(output_line)) # Print metrics print("Number of lines read: " + str(i_entries)) print("Number of comments: " + str(i_comments)) print("Number of entries: " + str(i_accepted_entries)) print("Number of duplicate entries: " + str(i_duplicate_entries)) print("Number of entries written: " + str(i_written_lines)) if __name__ == "__main__": # Parse arguments prsr_arguments = argparse.ArgumentParser(prog='gtf_to_position_file.py', description='Convert a GTF file to a positional file.', formatter_class=argparse.ArgumentDefaultsHelpFormatter) # Add positional argument prsr_arguments.add_argument("input_gtf", metavar="input_gtf", help="Path to the input GTF file.") prsr_arguments.add_argument("--attribute_name", metavar="attribute_name", default="gene_id", help="The name of the attribute in the GTF attributes to use instead of gene name, for example 'gene_name' or 'transcript_id'.") prsr_arguments.add_argument("output_positional", metavar="output_positional", help="Path for the output positional file.") args = prsr_arguments.parse_args() # Run Script convert_to_positional_file(args.input_gtf, args.output_positional, args.attribute_name) |
【本文地址】