单细胞层面的表达量差异分析到底如何做 |
您所在的位置:网站首页 › r语言如何做预测 › 单细胞层面的表达量差异分析到底如何做 |
单细胞层面的表达量差异分析到底如何做
|
差异分析当之无愧的是最常见的数据分析手段了,无论是转录组测序还是表达量芯片或者是蛋白质代谢组技术,最后都会得到数值矩阵。一般来说行是基因或者蛋白质等特征,这个可以很多,成百上千甚至过万。而列通常是样品,样品就有有表型,如果是两分组就自然而然的做差异分析了。大家手上都是有成熟的流程和代码,其实也就是跑了一下代码,十几分钟的事情。如果是需要理解图表的生物学含义,就需要看我六年前的表达芯片的公共数据库挖掘系列推文即可; 解读GEO数据存放规律及下载,一文就够解读SRA数据库规律一文就够从GEO数据库下载得到表达矩阵 一文就够GSEA分析一文就够(单机版+R语言版)根据分组信息做差异分析- 这个一文不够的差异分析得到的结果注释一文就够现在单细胞技术的大行其道,让我们的表达量矩阵里面的列也变成了成百上千甚至过万了,首先它们属于不同的样品,如果是10x技术的单细胞转录组,每个样品可以有5到8千的单细胞列,其次呢我们通常是做多个单细胞样品,详见:2个分组的单细胞项目标准分析,这样的话不同样品也可以有表型分组,比如处理组和对照组。 拿到了一个单细胞表达量矩阵,默认需要进行: 单细胞聚类分群注释 ,如果你对单细胞数据分析还没有基础认知,可以看基础10讲: 01. 上游分析流程02.课题多少个样品,测序数据量如何03. 过滤不合格细胞和基因(数据质控很重要)04. 过滤线粒体核糖体基因05. 去除细胞效应和基因效应06.单细胞转录组数据的降维聚类分群07.单细胞转录组数据处理之细胞亚群注释08.把拿到的亚群进行更细致的分群09.单细胞转录组数据处理之细胞亚群比例比较找各个单细胞亚群特异性高表达量基因(FindAllMarkers函数)我们以大家熟知的pbmc3k数据集为例,大家先安装这个数据集对应的包,并且对它进行降维聚类分群,参考前面的例子:人人都能学会的单细胞聚类分群注释 ,而且每个亚群找高表达量基因,都存储为Rdata文件。标准代码是: library(SeuratData) #加载seurat数据集 getOption('timeout') options(timeout=10000) #InstallData("pbmc3k") data("pbmc3k") sce FindMarkers函数结果也就是说,虽然我们是想找两个分组的差异,但是FindMarkers函数仍然是会优先得到样品特异性高表达量基因,因为在单个样品高表达量那么就很合理在样品所在的分组是高表达量的。 但是呢,实际上我们并不是需要这样的基因列表,它其实是样品特异性的,严格意义上来说这个就是批次效应,个体差异而已。之前我们在《单细胞天地》公众号分享过一个文献 ,解读在:https://cloud.tencent.com/developer/article/1901064 文章题目:Confronting false discoveries in single-cell differential expression日期:2021-09-28期刊:Nature Communications链接:https://www.nature.com/articles/s41467-021-25960-2里面提到的目前主流的单细胞差异分析方法都是Wilcoxon rank−sum test,但是它其实表现还不如pseudobulks 的方法: 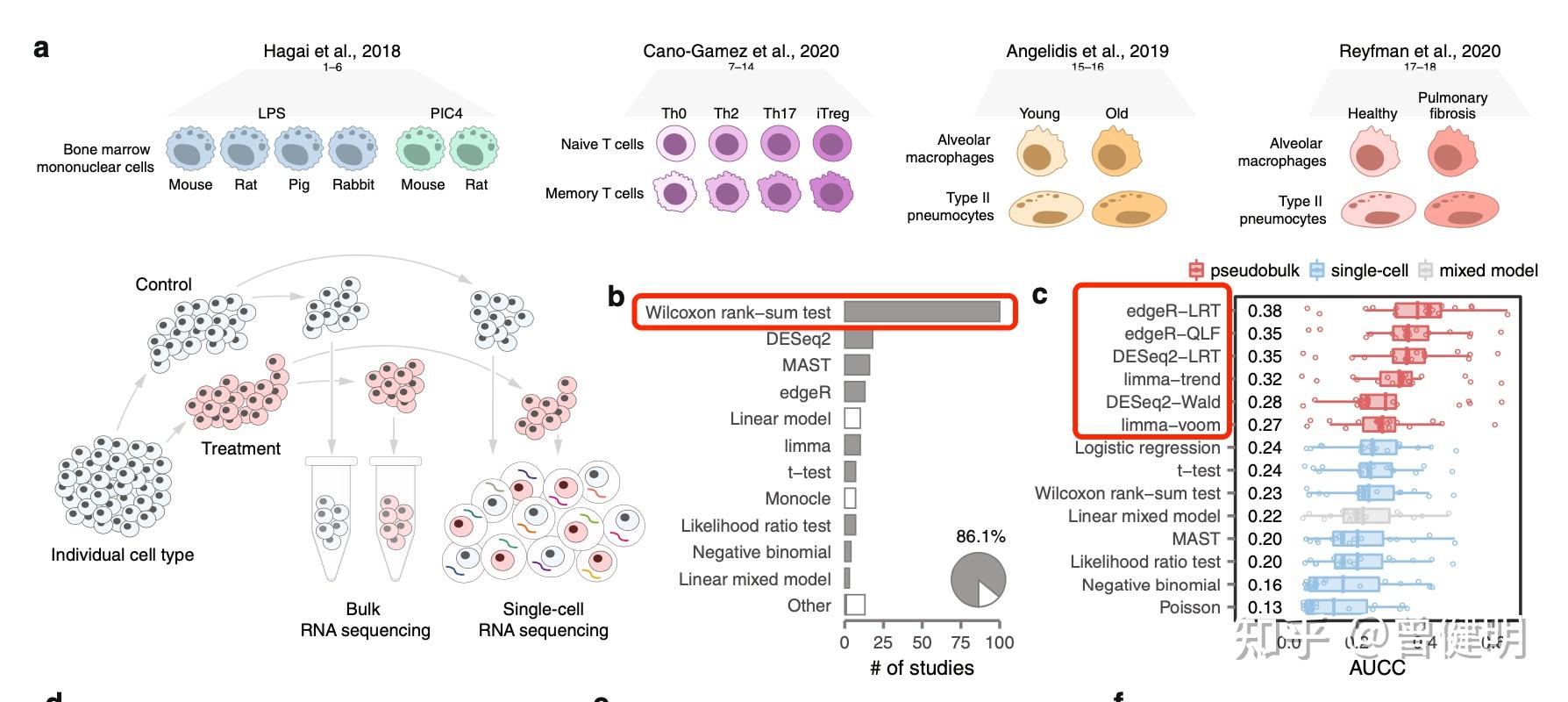 主流的单细胞差异分析方法都是Wilcoxon rank−sum test 主流的单细胞差异分析方法都是Wilcoxon rank−sum test为什么主流的单细胞差异分析方法都是Wilcoxon rank−sum test呢,因为大家都是使用(FindAllMarkers函数)或者(FindAllMarkers函数),你看看这两个函数的帮助文档: FindAllMarkers( object, assay = NULL, features = NULL, logfc.threshold = 0.25, test.use = "wilcox", slot = "data", min.pct = 0.1, min.diff.pct = -Inf,其中 test.use 参数非常重要,如下所示:  test.use 参数 test.use 参数默认的是就是 Wilcoxon Rank Sum test (default) 实现方法pseudobulks前面的 compSce是一个seurat对象 ,它里面的comp是表型是两个分组,然后mice是表型是十几个小鼠。也就是说十几个小鼠各自的单细胞转录组样品是两分组,需要做差异分析。 bs = split(colnames(compSce),compSce$mice) names(bs) ct = do.call( cbind,lapply(names(bs), function(x){ # x=names(bs)[[1]] kp =colnames(compSce) %in% bs[[x]] rowSums( as.matrix( compSce@assays$RNA@counts[, kp] )) }) ) phe = unique([email protected][,c('mice','comp')]) phe # 每次都要检测数据 group_list = phe[match(names(bs),phe$mice),'comp'] table(group_list) exprSet = ct exprSet[1:4,1:4] dim(exprSet) exprSet=exprSet[apply(exprSet,1, function(x) sum(x>1) > 1),] dim(exprSet) table(group_list) save(exprSet,group_list,file = 'input_for_deg.Rdata') load(file = 'input_for_deg.Rdata')有了表达量矩阵以及分组信息,后面就是常规的转录组差异分析啦。这里我比较喜欢 DESeq2 包: # 加载包 library(DESeq2) # 第一步,构建DESeq2的DESeq对象 colData |
【本文地址】
今日新闻 |
推荐新闻 |