单细胞分析实录(11): inferCNV的基本用法 |
您所在的位置:网站首页 › run三单怎么写 › 单细胞分析实录(11): inferCNV的基本用法 |
单细胞分析实录(11): inferCNV的基本用法
|
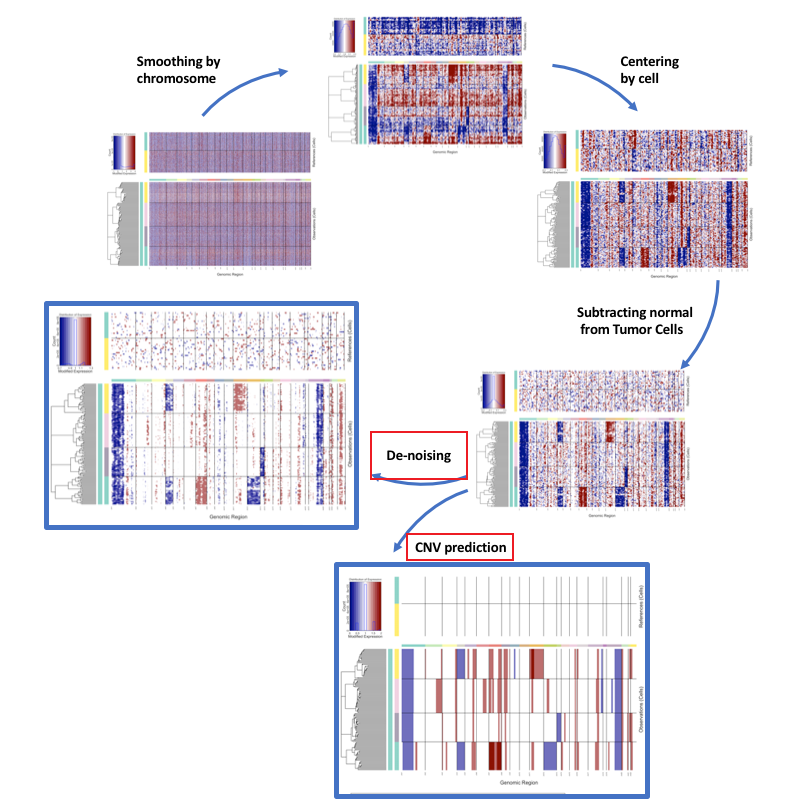
InferCNV可以用于肿瘤单细胞RNA-Seq数据中鉴定大规模染色体拷贝数变异,例如整个染色体或大片段染色体的扩增或缺失。基本思路是在整个基因组范围内,将每个肿瘤细胞基因表达与平均表达或“正常”参考细胞基因表达对比,确定其表达强度。 这段话来自InferCNV官方文档:https://github.com/broadinstitute/inferCNV/wiki 实际分析中,我们经常用inferCNV来判断肿瘤细胞。当然还可以做肿瘤异质性、克隆进化方面的探索,这部分我会在下一期的推文中介绍,本期推文介绍基本使用。公众号后台回复20210404获取今天的代码和测试数据。 1. 安装初次安装加载时,可能会提示你安装JAGS,根据提示安装JAGS后,再加载InferCNV就没问题 BiocManager::install("infercnv") library("infercnv") 错误: package or namespace load failed for ‘infercnv’: loadNamespace()里算'rjags'时.onLoad失败了,详细内容: 调用: fun(libname, pkgname) 错误: Failed to locate any version of JAGS version 4 The rjags package is just an interface to the JAGS library Make sure you have installed JAGS-4.x.y.exe (for any x >=0, y>=0) from http://www.sourceforge.net/projects/mcmc-jags/files 2. 输入在进行分析之前,需要准备三个文件: Raw Counts Matrix for Genes x Cells细胞的注释文件 共两列,一列CB,一列细胞来源,tab分割,无列名; 如果肿瘤细胞的注释类似malignant_{patient},则在后续聚类画图时,设置cluster_by_groups=T会根据patient的来源进行区分 该文件的CB可以小于count矩阵,此时inferCNV只会对这部分细胞进行分析;基因排序文件 tab分割,无列名 只有该文件和count矩阵共有的基因才会被分析(可以去掉不想画图的基因,比如线粒体基因),且该文件的染色体顺序决定了最终热图的染色体顺序(有些文章的图,inferCNV热图的染色体顺序是颠倒的,问题就出在这儿)。 我一般是根据cellranger提供的参考基因组gtf注释文件,得到基因排序文件 格式如下WASH7P chr1 14363 29806 LINC00115 chr1 761586 762902 NOC2L chr1 879584 894689 MIR200A chr1 1103243 1103332 3. 流程看一下这张流程图
我比较关注最后两步,去噪和CNV预测是分开的,一些已发表的文章这部分的分析都是基于去噪之后的结果(比如那张热图),CNV预测结果用的比较少,个人觉得使用analysis_mode = "subclusters"模式后的热图更好看,而且只有6个值 这是官网的一张对比图
主要步骤只有两步,如下 library(infercnv) #使用inferCNV之前,最好过滤掉低质量的细胞 infercnv_obj = CreateInfercnvObject(raw_counts_matrix="oligodendroglioma_expression_downsampled.counts.matrix", annotations_file="oligodendroglioma_annotations_downsampled.txt", delim="\t", gene_order_file="gencode_downsampled.EXAMPLE_ONLY_DONT_REUSE.txt", ref_group_names=c("Microglia/Macrophage","Oligodendrocytes (non-malignant)") ) #ref_group_names参数根据细胞注释文件填写,在示例中,这两种细胞是非恶性细胞,所以作为参照; #ref_group_names=NULL,则会选用所有细胞的平均表达作为参照,这种模式下,最好能确保细胞内在有足够的差异 infercnv_obj = infercnv::run(infercnv_obj, cutoff=1, out_dir="try", cluster_by_groups=TRUE, #analysis_mode="subclusters", #默认是"samples" denoise=TRUE, HMM=TRUE, num_threads=4 ) #去噪,HMM预测CNV这两项我一般都选上关于cutoff参数,官网是这样说的:cutoff=1 works well for Smart-seq2, and cutoff=0.1 works well for 10x Genomics,表示基因在所有参照细胞中,表达count的平均值的最小阈值,10X数据更稀疏,所以这个值小 cluster_by_groups:先区分细胞来源,再做层次聚类 out_dir表示输出文件夹的名字,没有会自动创建 5. 输出我认为重要的输出文件 infercnv.png降噪之后生成的最终热图,图中的数值是"Residual expression values",可以简单理解为基因表达值的另一种形式;
对应热图的参照panel infercnv.observations.txt : the tumor cell matrix data values对应热图的观测panel infercnv.observation_groupings.txtgroup memberships for the tumor cells as clustered,对应热图的观测panel的两个bar "Dendrogram Group" "Dendrogram Color" "Annotation Group" "Annotation Color" "MGH36_P9_B01" "1" "#8DD3C7" "1" "#8DD3C7" "MGH36_P3_E06" "1" "#8DD3C7" "1" "#8DD3C7" #2 3列只有一类值 #4 5列有四类值 infercnv.observations_dendrogram.txt树状图的newick格式,对应热图的观测panel的“树” run.final.infercnv_obj这个rds文件,以及包含在其中的三个子文件如下 @expr.data:对应最终热图的表达文件 @reference_grouped_cell_indices:对应最终热图的reference细胞名称 @observation_grouped_cell_indices:对应最终热图的observation细胞名称 HMM_CNV_predictions.*.pred_cnv_regions.dat:第二列是CNV的name,唯一; 第一列是CNV所属的group,inferCNV默认的模式(analysis_mode = "samples")是将一个patient作为一个整体找CNV,所以示例会有四个group; 4 5 6列包含CNV的坐标; 第三列表示状态: State 1: 0x: complete loss State 2: 0.5x: loss of one copy State 3: 1x: neutral State 4: 1.5x: addition of one copy State 5: 2x: addition of two copies State 6: 3x: essentially a placeholder for >2x copies but modeled as 3x $ less -S HMM_CNV_predictions.HMMi6.hmm_mode-samples.Pnorm_0.5.pred_cnv_regions.dat | head -n 3 cell_group_name cnv_name state chr start end malignant_MGH36.malignant_MGH36_s1 chr1-region_2 2 chr1 6281253 144341756 malignant_MGH36.malignant_MGH36_s1 chr1-region_4 3 chr1 151336778 156213123 HMM_CNV_predictions.*.cell_groupingstumor subclusters和cell的对应关系 只有analysis_mode = "subclusters"模式下才会生成,这个模式挺慢,但肿瘤异质性和克隆进化都是在这种模式下做的 cell_group_name cell malignant_MGH36.malignant_MGH36_s1 MGH36_P3_E06 malignant_MGH36.malignant_MGH36_s1 MGH36_P10_E07 HMM_CNV_predictions.*.pred_cnv_genes.dat每一个group(第一列), 每一个CNV片段(第二列)上面每一个基因(第四列)的CNV状态(第三列),文件中基因这一列是唯一的 $ less -S HMM_CNV_predictions.*.pred_cnv_genes.dat | head -n 3 cell_group_name gene_region_name state gene chr start end malignant_MGH36.malignant_MGH36_s1 chr1-region_2 2 ACOT7 chr1 6281253 6296032 malignant_MGH36.malignant_MGH36_s1 chr1-region_2 2 NOL9 chr1 6324329 6454451该文件可以查询感兴趣基因的CNV状态,如下: $ grep EGFR HMM_CNV_predictions.HMMi6.hmm_mode-samples.Pnorm_0.5.pred_cnv_genes.dat | head -n 3 malignant_MGH36.malignant_MGH36_s1 chr7-region_16 5 EGFR chr7 54819943 54827667 malignant_93.malignant_93_s1 chr7-region_98 4 EGFR chr7 54819943 54827667 malignant_97.malignant_97_s1 chr7-region_142 4 EGFR chr7 54819943 54827667这种方法看基因的CNV状态,也有局限性,只有当这个基因与它所处的CNV region的CNV状态比较一致才会被找出来,如果这个基因的CNV状态比周围一些基因的CNV状态强很多,则找不出来。 个人猜测是因为inferCNV是画窗口针对相邻的多个基因做推断,如果只有一个基因表达很高或很低,则被平均掉 上述的这些输出文件,对于深入分析CNV很有用,但是如果只区分肿瘤细胞,则用不到这么多文件。 6. 画图infercnv包也包含了画图函数plot_cnv,这个知道的人不多, library(RColorBrewer) infercnv::plot_cnv(infercnv_obj, #上两步得到的infercnv对象 plot_chr_scale = T, #画染色体全长,默认只画出(分析用到的)基因 output_filename = "better_plot",output_format = "pdf", #保存为pdf文件 custom_color_pal = color.palette(c("#8DD3C7","white","#BC80BD"), c(2, 2))) #改颜色
后续应该还有两篇inferCNV的内容,敬请期待~ 因水平有限,有错误的地方,欢迎批评指正! |



【本文地址】
今日新闻 |
推荐新闻 |