用R分析微生物组群落数据(1) 软件安装、数据集下载和导入 |
您所在的位置:网站首页 › rstudio怎么导入数据集 › 用R分析微生物组群落数据(1) 软件安装、数据集下载和导入 |
用R分析微生物组群落数据(1) 软件安装、数据集下载和导入
|
参考: https://grunwaldlab.github.io/analysis_of_microbiome_community_data_in_r/index.html 1.下载需要的软件和数据 1.1 安装R、Rstudio和必要的R包 1.1.1 安装RR是一门关注统计学、数据科学、可视化的编程语言。它可以再所有的公共操作系统安装,R语言可以从如下地址下载: https://cran.r-project.org/ 1.1.2 安装RStudioRStudio是一个交互式的开发环境( “Integrated Development Environment” ,IDE),即“带有额外编程相关工具的文本编辑器”。RStudio使得R用起来更简单,但是不依赖于R。大多数R语言用户都会使用RStudio, 因此我们推荐使用这个作为工作界面。 RStudio的主要版本是免费开源的,可以通过如下地址下载: https://www.rstudio.com/products/rstudio/download/#download 1.1.3 安装必要的R包除了R语言安装过程中包含的包,安装R包是很普遍的。 R语言包是以标准化方式打包到一起的工具的集合,很容易安装。实际上,R语言中的大多数功能是来自于R包,而非基础的R。下面的包将用在这个基础教程中。点击它们的名字就可以跳转到这个包的文档页面: vegan: 包括很多能够用于生态学研究的标准化统计工具。被phyloseq和metacoder等包广泛使用。(Dixon 2003)metacoder: 处理和可视化分类(taxonomic)数据,特别是来自扩增子宏基因组研究的。(Foster, Sharpton, and Grünwald 2017)taxa:定义分类 classes 以及操纵它们的函数。目标是用这些类型作为R包能够构建和使用的低水平基础分类类型。这个能够被metacoder使用。 (Foster, Chamberlain, and Grünwald 2018)phyloseq: 包含分析和可视化微生物组数据工具的流行包。 (McMurdie and Holmes 2013)ggplot2:漂亮的画图包。(Wickham 2009)dplyr:通过内聚和直观的命令集来操纵扁平数据的包。是基本R语言方法的一个流行的替代平。 (Wickham and Francois 2015)readr:使得从文件读取扁平数据更加简单。stringr:文本操纵的相关函数。agricolae:用于实验的设计和分析,特别是植物相关的实验。ape :用于DNA序列分析和进化的流行包。我们写和活跃地维护了metacoder(Foster, Sharpton, and Grünwald 2017) 和taxa(Foster, Chamberlain, and Grünwald 2018), 因此他们非常依赖于这个基本教程。 更多用于微生物组数据分析的R资源可以通过如下链接获取: https://microsud.github.io/Tools-Microbiome-Analysis/ 你可以在R语言编译器中输入下面的代码来安装这些包: # Install phyloseq from Bioconductor if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager") BiocManager::install() BiocManager::install("phyloseq") # Install the rest of the packages from CRAN install.packages(c("vegan", "metacoder", "taxa", "ggplot2", "dplyr", "readr", "stringr", "agricolae", "ape"), repos = "http://cran.rstudio.com", dependencies = TRUE)如果安装不成功,可以尝试安装内次安装一个包(如:install.packages("taxa")),然后查找错误信息。通常问题是需要安装一个非R依赖项,如何安装它取决于你的操作系统。将错误信息复制到Google通常可以帮你找出错误原因。如果一个包已经被正确安装,你应该能够用library来加载它(如:library(phyloseq))。 如果你想检测通过开源的Rstudio检查软件是否安装成功,运行下面的代码。如果你使用R或RStudio需要帮助,查看附件: library(metacoder) x = parse_tax_data(hmp_otus, class_cols = "lineage", class_sep = ";", class_key = c(tax_rank = "info", tax_name = "taxon_name"), class_regex = "^(.+)__(.+)$") heat_tree(x, node_label = taxon_names, node_size = n_obs, node_color = n_obs)
在这个workshop中,我们会使来自Wagner et al. (2016)的数据,一个探索植物年龄、基因型、环境对严格布氏小蠊(Boechera stricta,芥菜科多年生草本植物)的细菌群落的影响的研究。Wagner et al. (2016)发布了这篇文章的原始数据,它可以再在 dryad的此处获取。这是一个分享你的原始数据的好例子。 这个数据的副本已经包含在这个网站,能够通过下面的链接下载。 2.1 Sample metadatasample data是一个表格,每一行是一个样品,样品的信息在每一列,如:植物的基因型和产地。它是一个170kb的以tab分割的文本文件。 SMD.txt 2.2 The OTU abundance table这个表有每个样品的每个OTU的reads数目。它是一个6Mb压缩的以tab分割的文本文件。 otuTable97.txt.bz2 2.3 The OTU taxonomy file这个文件有每个OTU的taxonomic classifications,它是一个6Mb的tab键分割的文本文件。 taxAssignments97.txt 3. 将数据输入R 3.1 典型的微生物组数据集大多数处理高通量amplicon数据的pipeline,例如mothur, QIIME和dada2, 都会产生一个read数目的矩阵。矩阵的一个维度(行或者列)由 Operational Taxonomic Units (OTUs), phylotypes, or exact sequence variants (ESVs)(所有都是二进制的相似读段序列) 组成。另一维由samples组成。不同的工具将期望或者输出不同方向的矩阵,但是在我们的案例中,列是samples,行是OTUs。有时候OTU数据矩阵丰度是两个分开的表格。通常有另外一个行是样本信息的表格。这使得增加很多额外的可作为子集数据的样本数据列更加简单。每一个样本和OTU都有独一无二的ID。 如果数据已经格式化得很好了,导入数据到R是很简单的,否则可能是一个令人沮丧的过程。 .csv (comma-separated value)或.tsv (tab-separated value)是格式化好的数据,每一个都是一个单独的表格,没有额外的评论或者形式(如:合并的单元格)。这些格式化的文件都可能有.txt后缀(后缀不是真正重要的,它是针对人的,不是针对计算机的)。正确的数据格式化的更多信息,可以看我们的重现研究指南的数据格式化小节。只要可能,您应该始终导入原始输出数据,并避免任何“手动”操作(如:non-scripted)去修改数据,特别是Excel程序,可能会不时地篡改数据 (Zeeberg et al. (2004))。 在这个workshop中,我们将使用来自Wagner et al. (2016)的数据,一个探索植物年龄、基因型、环境对严格布氏小蠊(Boechera stricta,芥菜科多年生草本植物)的细菌群落的影响的研究。这里是由Mary Ellen Harte拍摄的Boechera stricta的照片: 
Wagner et al. (2016)发布了这篇文章的原始数据,它可以再在 dryad的此处获取。这是一个分享你的原始数据的好例子。 有很多functions可以用来读取扁平化数据,包括base R的方法,如read.table和read.csv,但是我们将使用readr package中的函数,其返回tibbles而不是data.frame(R语言中的“table”)。 library(readr) # Loads the readr package so we can use `read_tsv`Tibbles是一种具有更优美输出和更一致表现的data.frame。点击此处下载OTU表。让我们先读取原始的OTU表: otu_data |
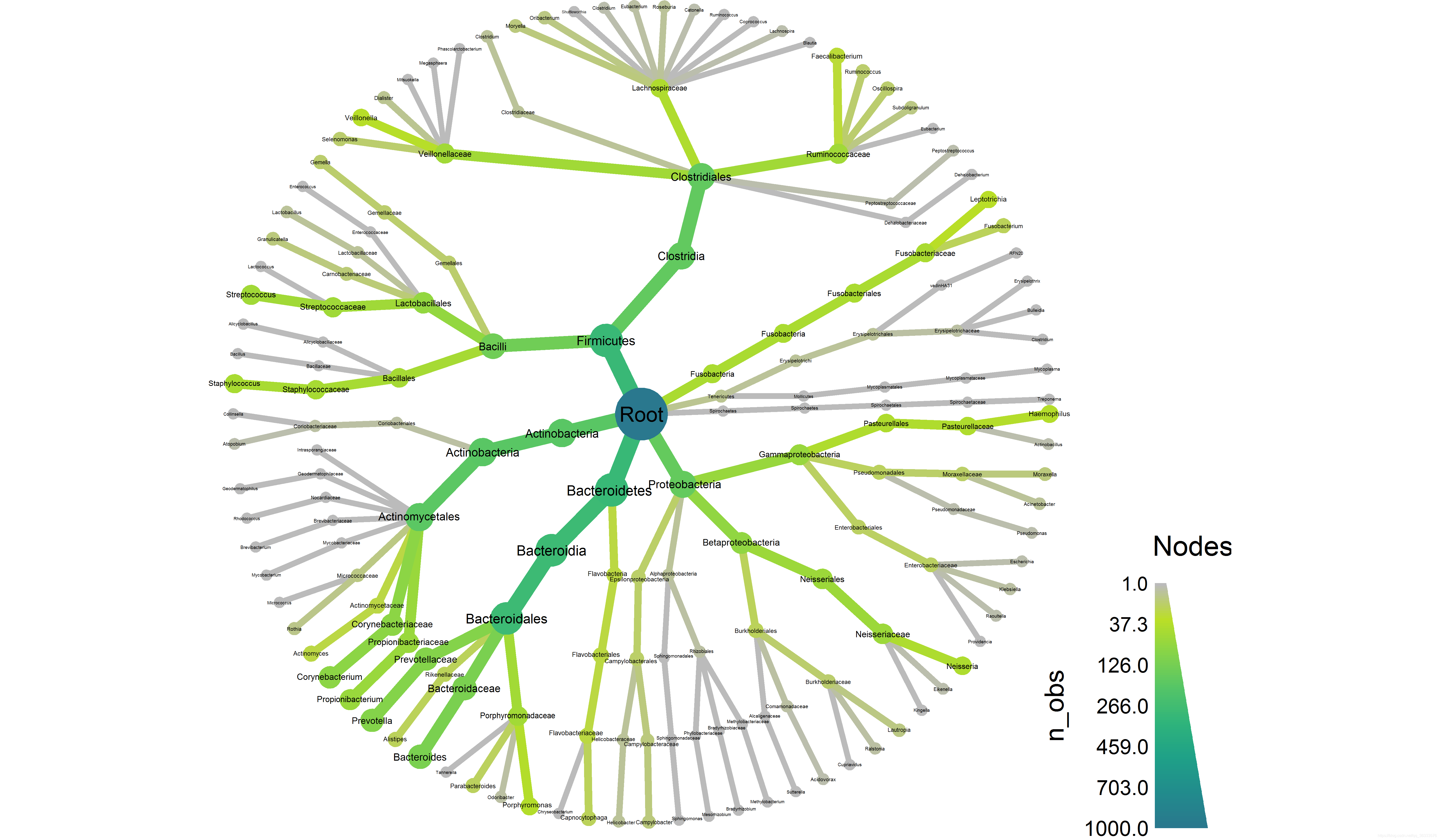 如果你看到了上面的图片,那么祝贺你。你现在应该已经获得这个工作所需的使用R的所有集合。如果你不能够看到这个图,那么请将下面代码运行的结果发邮件给我们,帮助我们排除这个问题的故障。
如果你看到了上面的图片,那么祝贺你。你现在应该已经获得这个工作所需的使用R的所有集合。如果你不能够看到这个图,那么请将下面代码运行的结果发邮件给我们,帮助我们排除这个问题的故障。
【本文地址】
今日新闻 |
推荐新闻 |