小白轻松入门GWAS:从数据准备到可视化结果 |
您所在的位置:网站首页 › qq收集表结果在哪看 › 小白轻松入门GWAS:从数据准备到可视化结果 |
小白轻松入门GWAS:从数据准备到可视化结果
|
在群体重测序的文章中,我们经常看到利用GWAS筛选候选基因。 那么,什么是GWAS?GWAS如何进行分析?需要准备哪些数据?有哪些可视化结果?这些都是新人常常疑惑的问题。本期,我们将与大家探讨全基因组关联分析(GWAS)。 GWAS是一种统计学的研究方法。它可用于在许多不同人群中识别与特定疾病或生物学特征相关的基因变异。这种方法通过比较患有特定疾病的个体与不患病的个体的基因组,来寻找与疾病风险相关的基因标记,特别适合复杂疾病。而在农业和生物多样性研究中,GWAS被用来识别影响作物产量、抗病性、生长速度以及其他农艺性状的基因。此外,GWAS也应用于动物,帮助我们理解疾病机制和遗传性状,这对于畜牧业和宠物繁育同样有重要价值。
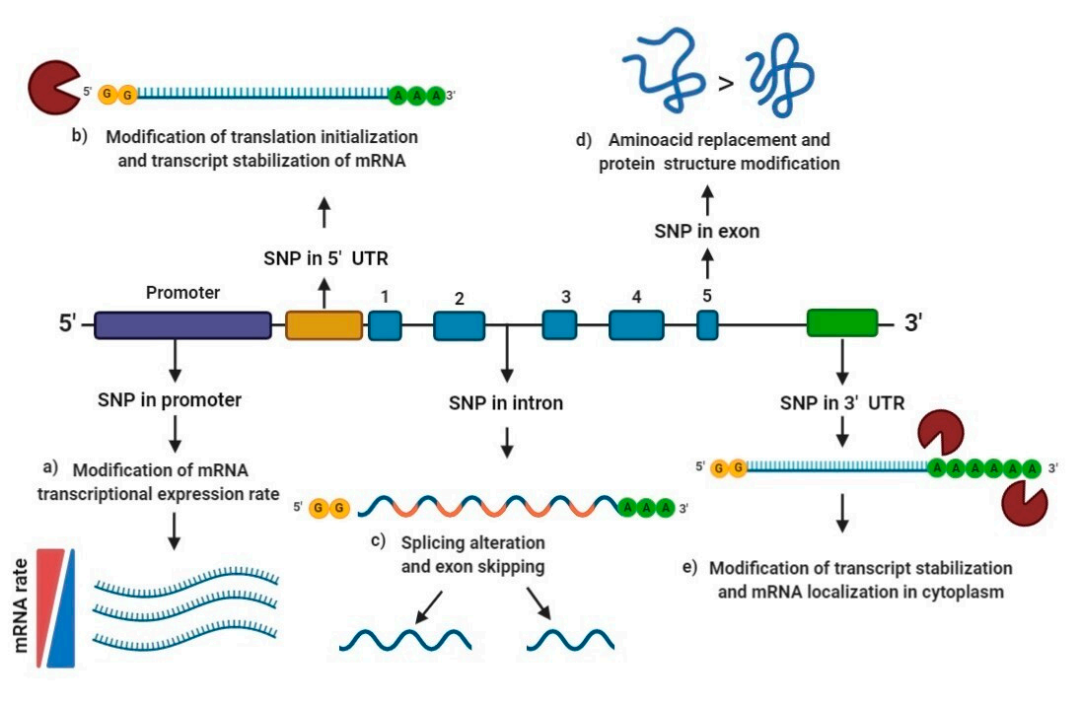
应用方向 在重测序分析中,将测序数据比对到参考基因组上后,会进行变异检测;而变异类型主要有单核苷酸变异(SNP)、插入缺失(Indel)、拷贝数变异(CNV)和结构变异(SV)等变异类型。GWAS是基于SNP的关联分析,所以我们先了解下SNP。 SNP的概念 单核苷酸多态性(Single Nucleotide Polymorphisms,SNPs)是指在基因组的特定位置上,单个核苷酸(A、T、C或G)发生变异,导致不同个体之间在该位置上的碱基不同。这种变异是最常见的遗传多态性形式,大约平均每1000个碱基中就有一个多态位点。 假设我们有两个DNA序列: 参考序列:...AAGCTATGC... 变异序列:...AAGCTGTGC... 在这个例子中,参考序列中的碱基“A”被变异序列中的碱基“G”所取代,这就是一个SNP。SNPs可以出现在基因组的任何位置,包括编码区、非编码区、调控区等。根据其位置和性质,SNPs可以分为以下几类: a.启动子区域的SNPs: 通过改变转录因子结合位点的构象来调节基因表达; b.5’UTR区域的SNPs: 可能改变信使核糖核酸(mRNA)的翻译初始化和转录稳定性; c.内含子区域的SNPs: 可能产生剪接改变、外显子跳跃,并调节核输出、转录速率和转录稳定性; d..外显子区域的SNPs: 可能导致一个氨基酸取代另一个氨基酸,也称为非同义多态性,这可能产生蛋白质结构修饰;外显子区域除了非同义多态性,还有同义编码SNP,同义编码SNP本身不会改变蛋白质序列,因为并不是所有密码子的改变都会改变氨基酸序列,但这并不意味着这部分的SNP就对表型没有影响。蛋白质翻译存在密码子偏好性,也有可能会导致表型变化。 e.3’UTR中的SNPs: 可能改变转录稳定和mRNA的定位。
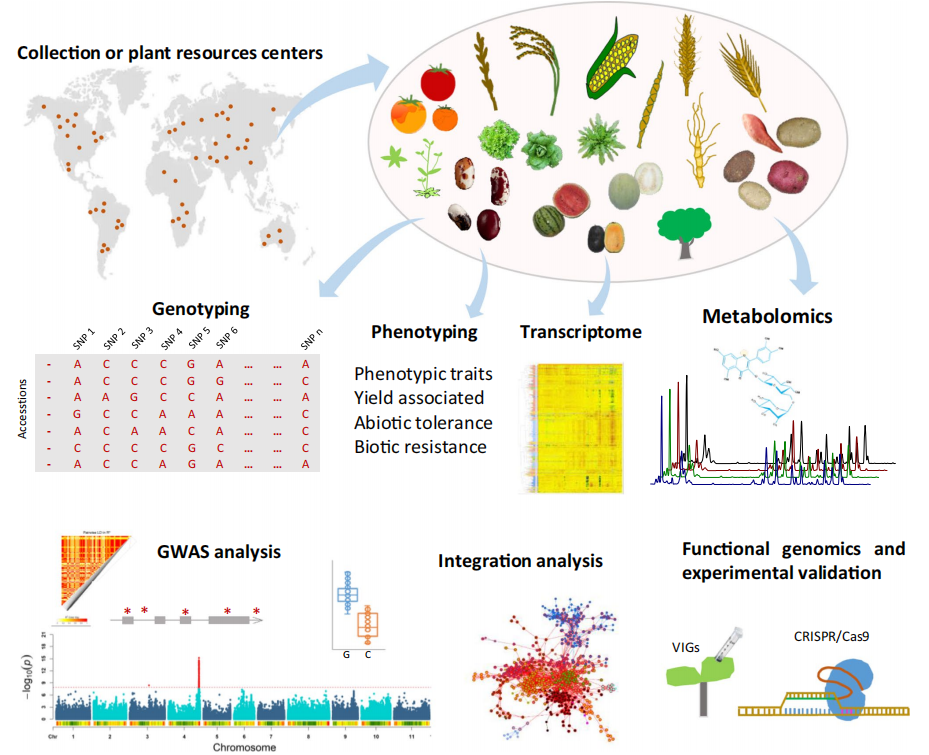
图:单核苷酸多态性(SNPs)根据其位置而产生的功能效应[1] SNPs是基因组中最常见的遗传变异形式,通过影响蛋白质功能、基因表达等机制,对个体的表型产生重要影响。研究SNPs具有广泛的意义,不仅在医学领域推动疾病研究和个性化医疗的发展,还在农业领域促进作物和动物育种、提高生产效率、保护遗传多样性等方面发挥重要作用。同时,SNPs研究在进化生物学、生态学和群体遗传学等基础研究领域也具有重要价值。 了解完SNP,我们再来看看GWAS的分析流程,它是如何将SNP信息和表型数据结合起来的,寻找关联位点。 GWAS流程 以植物为例,GWAS流程如下: (1) 选择合适的群体,收集表型信息 (2) 高通量测序,基因分型 (3) GWAS关联分析 (4) 整合分析 (5) 候选基因挖掘及功能验证
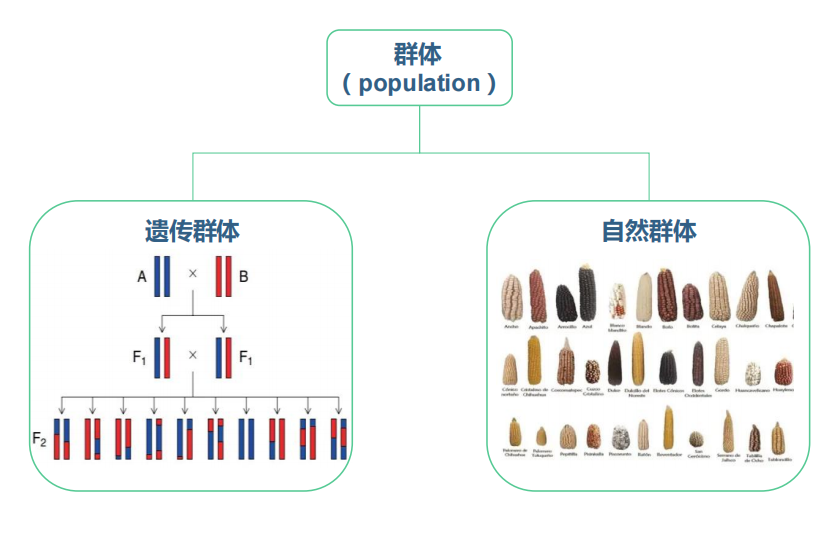
图:植物中GWAS的流程示意图[2] 群 体在农学上,研究要求较大的样本规模,以确保统计效力,并且样本应具有良好的代表性,覆盖目标群体的遗传多样性。常见的群体有自然群体、遗传群体。
选择合适的群体后,我们需要准备两个数据:表型数据和基因型数据。 表型数据谈起表型数据,我们先来了解下GWAS的表型性状。 GWAS中的表型性状可以分为三类:数量性状、质量性状和分级性状。 01 数量性状(Quantitative Traits) 数量性状是指可以用数字值来描述的性状,例如: 身高(cm) 体重(kg) 籽粒数(个) 产量(kg/亩) 这些性状可以通过测量获得连续的数字值,通常遵循正态分布或近似正态分布。一般由多基因控制,能够测量得到具体数值,受环境影响大。所以,尽量保证样本材料在相同或尽可能一致的环境条件下培育或养殖。这样的控制可以帮助我们更精准地识别出基因对性状的影响,减小环境变量带来的噪音。 02 质量性状(Qualitative Traits) 与数量性状相反,其无法用固定数值表示,而是表现出一种状态,例如: 花色(红、黄、白等) 果实形状(圆、椭圆等) 疾病(有或无) 质量通常是由一个或少数几个基因控制的,表现为离散的、可区分的类别,例如豌豆的花色或人类的血型。这类性状无法用具体数值衡量,但可以用分类变量表示,比如用0、1等标识不同类别。为了确保分析的准确性和统计效能,建议在采集样本时尽量保证各类别的样本数量相近。 03 分级性状(Ordinal Traits) 分级性状是介于质量性状和数量性状之间的一类性状,表现为有序的类别,但这些类别之间的差异不是连续的。例如,抗病性可以分为低、中、高三类。分级性状通常由多基因控制,并且可能受到环境因素的影响。例如: 病毒抵抗性水平(高、中、低) 籽粒颜色(浅黄、深黄、棕色等) 植株高度级别(高、中、低) 这些性状可以用数字值(0、1、2等)来描述,但具有明确的等级或顺序,比如将疾病严重程度分为轻度(1)、中度(2)和重度(3)。 表型数据通常是一个数据框(如下图),行表示个体样本,列表示不同的表型。举个例子:选取了1000个小麦样本作为研究对象,这些样本来自不同的品种和地理位置。我们关注的是小麦的株高性状,这是一个数量性状,可以用厘米来衡量。 表型数据参考模板如下:
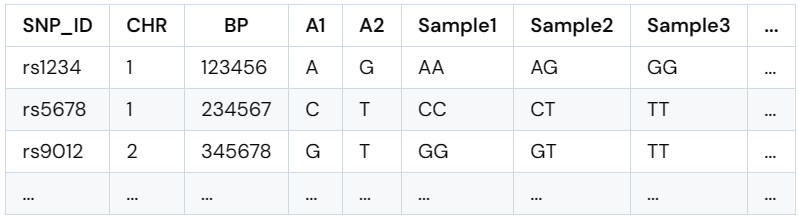
表型数据参考模板 基因型数据基因型是指一个个体在某个特定基因位点上所拥有的等位基因的组合。每个基因位点可以有不同的等位基因,这些等位基因是由父母各自传递的一个单倍体组成。
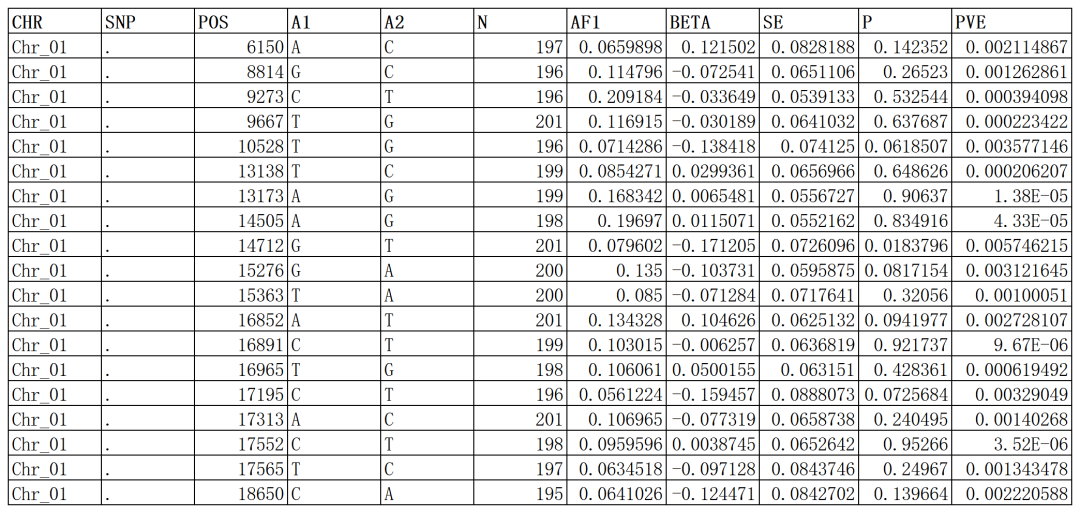
基因型数据的参考模板 SNP_ID:SNP标识符,通常以"rs"开头,后跟一个唯一的数字。 CHR:染色体编号,表示SNP所在的染色体。 BP:碱基对位置,表示SNP在染色体上的位置。 A1:等位基因1,表示SNP的参考等位基因。 A2:等位基因2,表示SNP的替代等位基因。 Sample1,Sample2,Sample3,...:每个样本的基因型,使用两个等位基因的组合表示(如AA,AG,GG)。 如何计算GWAS数据 前期数据准备好,我们就需要利用相关软件(比如PLINK或GCTA等,我们使用的是GCTA)进行GWAS分析,找到与目标表型相关的候选位点。GWAS的初步结果通常会给出一个表格结果,如下图:
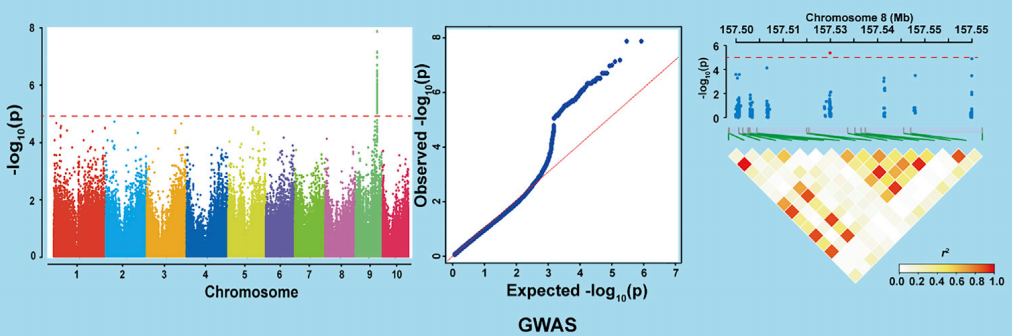
GWAS结果 CHR:染色体编号,通常用阿拉伯数字表示。 SNP:单核苷酸多态性,也称为遗传变异,没有通用名留空。 POS:基因组物理位置,以碱基对(bp)为单位。 A1:SNP上的第一个等位基因,通常选择出现频率较高的那个。 A2:SNP上的第二个等位基因,通常选择出现频率较低的那个。 N:属于该种类型等位基因的样本数 AF1:SNP上的第一个等位基因频率 BETA: 拟合参数 SE:每个等位基因贡献效应值的标准误差 P:显著性水平,表示该SNP与性状是否相关的统计显著性程度,通常以科学计数法表示。 PVE:该位点表型变异解释百分比。 结果表格中列出来一些结果:染色体信息、SNP、基因组位置、等位基因信息、出现等位基因的样本数、第一个等位基因频率,还有相应的P值(通过统计检验计算)。我们主要关注P值。P值(p-value)是一个重要的统计指标,用于评估某个基因变异(通常是单核苷酸多态性,SNP)与研究的性状或疾病之间关联的显著性。P值越小,表示SNP与表型的关联性越强。 GWAS可视化结果 GWAS中有三个非常典型的可视化结果图:曼哈顿图、QQ图和LD-Block图。
GWAS可视化结果[2] Manhattan图因其形似曼哈顿摩天大楼,故俗称为曼哈顿图。本质上它是一个散点图,一种用于展示GWAS结果的常用可视化工具。在曼哈顿图中,横轴表示基因组的染色体位置,按照染色体的顺序排列,每个染色体用不同的颜色表示。纵轴表示-log10(P),通常越高表示关联越显著。因此,曼哈顿图的纵轴可以帮助研究人员快速识别基因组中具有显著关联的区域。 图中还会设置一条阈值线(下图虚线,通常设置为P |

【本文地址】
今日新闻 |
推荐新闻 |