多个基因型数据文件如何高效合并 |
您所在的位置:网站首页 › plink使用 › 多个基因型数据文件如何高效合并 |
多个基因型数据文件如何高效合并
|
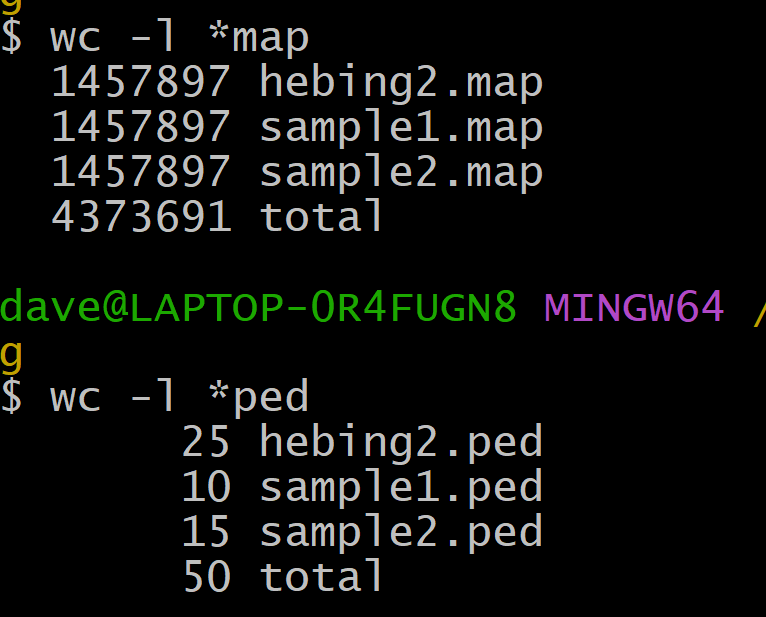
大家好,我是邓飞,这里总结一下多个plink文件合并的问题。 合并有两种应用场景: 1,样本一样,位点不一样,不如同样的样本,第一号染色体的数据,第二号染色体的数据合并。 2,位点一样,样本不一样,比如同样的芯片数据(map数据一样),第一批的数据,第二批的数据。 所以,这里也分为两种方法总结一下。 1. 样本一样,位点不一样典型的情况:现在有4条染色体的数据,每个染色体一套plink文件,如何合并在一起。 比如数据: dat_chr_1.mapdat_chr_2.mapdat_chr_3.mapdat_chr_4.map dat_chr_1.peddat_chr_2.peddat_chr_3.peddat_chr_4.ped这里使用--merge-list,对多个文件进行合并。 首先,我们先生成一个txt文件,把需要合并的ped和map数据的名称放进去,ped在前面,map在后面。 下面的文件名为:p12.txt,分为两列内容,第一列为ped的名称,第二列为map的名称,每一行都是一对plink文件。 dat_chr_1.peddat_chr_1.map dat_chr_2.peddat_chr_2.map dat_chr_3.peddat_chr_3.map dat_chr_4.peddat_chr_4.map代码如下: plink--merge-listp12.txt--recode--outhebing日志如下行: $plink--merge-listp12.txt--recode--outhebing PLINKv1.90b6.2164-bit(19Oct2020)www.cog-genomics.org/plink/1.9/ (C)2005-2020ShaunPurcell,ChristopherChangGNUGeneralPublicLicensev3 Loggingtohebing.log. Optionsineffect: --merge-listp12.txt --outhebing --recode 15236MBRAMdetected;reserving7618MBformainworkspace. Performingsingle-passmerge(165people,426095variants). Mergedfilesetwrittentohebing.bed+hebing.bim+hebing.fam. 426095variantsloadedfrom.bimfile. 165people(80males,85females)loadedfrom.fam. 112phenotypevaluesloadedfrom.fam. Using1thread(nomultithreadedcalculationsinvoked). Beforemainvariantfilters,112foundersand53nonfounderspresent. Calculatingallelefrequencies...done. Totalgenotypingrateis0.997722. 426095variantsand165peoplepassfiltersandQC. Amongremainingphenotypes,56arecasesand56arecontrols.(53phenotypes aremissing.) --recodepedtohebing.ped+hebing.map...done.结果文件: map数据之和,是合并后的map数据。 $wc-l*map 119487dat_chr_1.map 119502dat_chr_2.map 98971dat_chr_3.map 88135dat_chr_4.map 426095hebing.map 852190totalped数据不变: $wc-l*ped 165dat_chr_1.ped 165dat_chr_2.ped 165dat_chr_3.ped 165dat_chr_4.ped 165hebing.ped 825total 2. 位点一样,样本不一样同样使用上面的方法。用--merge-list,然后定义名称的文件去进行合并。 这里用两个plink文件,sample1和sample2,多个文件操作方法是一样的。 sample1.mapsample1.pedsample2.mapsample2.ped生成p12.txt文件: sample1.pedsample1.map sample2.pedsample2.map运行命令合并: plink--merge-listp12.txt--recode--outhebing2日志如下: $plink--merge-listp12.txt--recode--outhebing2 PLINKv1.90b6.2164-bit(19Oct2020)www.cog-genomics.org/plink/1.9/ (C)2005-2020ShaunPurcell,ChristopherChangGNUGeneralPublicLicensev3 Loggingtohebing2.log. Optionsineffect: --merge-listp12.txt --outhebing2 --recode 15236MBRAMdetected;reserving7618MBformainworkspace. Performingsingle-passmerge(25people,1457897variants). Mergedfilesetwrittentohebing2.bed+hebing2.bim+hebing2.fam. 1457897variantsloadedfrom.bimfile. 25people(13males,12females)loadedfrom.fam. 17phenotypevaluesloadedfrom.fam. Using1thread(nomultithreadedcalculationsinvoked). Beforemainvariantfilters,17foundersand8nonfounderspresent. Calculatingallelefrequencies...done. Totalgenotypingrateis0.996107. 1457897variantsand25peoplepassfiltersandQC. Amongremainingphenotypes,10arecasesand7arecontrols.(8phenotypesare missing.) --recodepedtohebing2.ped+hebing2.map...done. Warning:2het.haploidgenotypespresent(seehebing2.hh);manycommands treattheseasmissing.「结果如下:」 map数据完全一样,ped数据相加。 「注意1:如果位点不一样,会计算两个map的并集」 「注意2:合并时,不是根据染色体+物理位置,而是根据第二列map的名称,要确保有交集,否则合并的结果是错误的」 「注意3:样本合并时,如果样本ID有重复,会报错。建议提取检验」 最后推荐一个GWAS分析教程,干货满满,值的学习: GWAS分析先做后学(点击查看教程) |
【本文地址】
今日新闻 |
推荐新闻 |