有机小分子化合物解离常数pKa的预测研究进展 |
您所在的位置:网站首页 › pka与酸碱性的关系 › 有机小分子化合物解离常数pKa的预测研究进展 |
有机小分子化合物解离常数pKa的预测研究进展
|
其中, A 代表酸分子解离后形成的共轭碱. 解离常数定义为该化学反应的平衡常数, 假定相关物质的活 度可以用浓度数据代替且水分子浓度为常数, 则:
鉴于Ka的数量级, 常使用负对数形式来表示酸解离常数:
因此, pKa为50%化合物发生解离时溶液的pH. pKa越小, 化合物酸性越强. 通常情况下, 水中弱酸 pKa的范围为2~12. 相应地, 碱的解离常数使用其 共轭酸的pKa来表示 :
1.2 pKa 的热动力学背景 水溶液中, 酸AH中的质子转移到水分子上:
这个过程受反应的自由能控制:
通过热动力学平衡常数K, 可以建立Gaq与解离 常数pKa之间的线性相关:
对应气相中的反应表示为
对于每个物种, 溶液中的自由能都可以分解成 气相自由能与溶解自由能之和, Gaq(AH)=G (AH)+ Gs (AH), 则整个质子反应过程:
因此,
式(6)~(8)提供了一种pKa的理论计算方法, 即在不同的量子化学水平上计算气相质子转移能G 与溶解自由能Gs , 进而得到Gaq与pKa. 在气相和水相中的质子转移过程与反应自由能和溶解自由能的关系如图1所示.
1.3 含多个可解离官能团的化合物的pKa 如果化合物含有n 个可解离的官能团, 溶液体系中总共可形成2n 个微物种(考虑到每个官能团解离与 否, 共有2n 种组合)和n2n−1个micro-pKas(2个微物种之 间的平衡常数). 所有带有相同数目的未解离质子的 微物种组成了体系的宏观状态, 共有n+1个(分别有0, 1,···, n个未解离质子), 即产生n个macro-pKas. 图2以 西替利嗪为例展示了带有3个可解离官能团的化合物 的解离过程, 表1列出了micro-pKas的实验测定值. 当 n>3, 如果没有额外的信息或假设条件, micro-pKas无法通过滴定曲线获得[9].
图 2西替利嗪的解离图解. 西替利嗪有3个解离官能团, 因此有 23 =8 个微物种和 3×22 =12 个 micro-pKas, 以及 3+1=4 个宏观状态和 3 个 macro-pKas. h 代表结合的氢原子个数[8]
2.1 数据来源与可获得性 到目前为止, 已有文值, 例如, Beilstein数据库整合了很多小分子化合物pKa实验测定的文献, 共包含约15 万个pKa数据, 可以通过MDL Crossfire Commandar获得[11]; Lange's化学手册也是pKa实验数据的一个重要来源[12]. 还可以使用一些商业软件, 比如ACD和 SPARC, 查询化合物的pKa实验值, 其中, SPARC仅 能提供与所查询的化学结构完全一致的物质pKa数据[13], 而ACD允许用户进行相似性定义, 除了能给出精确匹配的物质, 还能提供一些结构相似物质的 pKa值[14]. 表S1总结了最新的免费和商业pKa数据库 以及包含pKa数据的书籍[15~33]. 2.2 数据质量 除了数据来源(很多数据库是商业性质的), 数据 质量是另外一个重要问题. 即使是公开发表的数据, 也不一定都可靠, 有些时候甚至相互矛盾. 例如, 在 Beilstein数据库中搜索苯酚, 会得到近30个数据, 并 呈现2个峰值, 有6个数据在1.0左右, 另外20几个数据分布在10.0左右. 很显然, 1.0是错误数据, 是再 次对pKa的负对数[34]. 可接受的实验测定误差大概为0.5个pKa [35]. 除 了实验本身的误差或者错误以外, 数据集的质量问 题还来自于以下几个方面[8]: (1) pKa数据被分配给错 误的化合物, 导致这个问题的原因可能是化合物名字有歧义、不标准, 或者是印刷错误等; (2) 错误的数值, 比如上面苯酚的例子, 或者pKa数据的印刷错误、 使用Ka或pKb(碱)代替pKa等; (3) 对含多个可解离中 心的化合物, pKa分配错误; (4) 同1个物质数据重复 输入, 即使数据是完全相同的, 在训练集中重复出现 会导致该化合物的重要性提高, 如果同时出现在训 练集和预测集中, 会提高验证结果的统计学性能; (5) 使用预测值代替实验值; (6) 不一致的实验条件, 比 如温度或者溶剂; (7) 不同实验室或实验人员的误差, 例如对二氯苯二磺胺, 有不同的文献数据, pKa1= 8.24, pKa2=9.50[36] vs. pKa1=7.40, pKa2=8.60[37]. 基于上述原因, 在使用这些数据时, 特别是用于 开发预测模型时, 要进行前处理, 例如筛选实验测定 条件、不同来源的数值进行统计分析、比照系列物的 pKa值、查阅原始文献等, 这些处理都会在一定程度上减少或消除数据质量问题. 2.3 pKa实验测定方法 现代分析化学的发展提供了多种pKa测定方法, 有些已经实现自动化操作. 常用的实验手段包括电位滴定法、电导滴定法、分光光度法、毛细管电泳法 (CE)、核磁共振法(NMR)、高效液相色谱法(HPLC), 这些方法也可以联合使用进行pKa的高通量筛选, 例 如CE与质谱结合、HPLC与质谱结合等. 各种方法的 测定原理及优缺点, 详见文献[34, 38]. Lee和Crippen[34]分析了Beilstein数据库中的pKa, 发现近一半的数据未提及测定方法, 另外一半数据, 绝大多数是使用电位滴定法、分光光度法和电导测定 法得到的, 占总量的98%以上. 此外, 尽管该数据库 包含了近15万pKa数据, 但对于单解离中心的化合物, 去除重复、可疑数据之后, 可靠的pKa数据不足2000个[5]. 由此可见, 作为化合物最基础的理化性质参 数, pKa的实验数据还相当缺乏, 并且存在着准确性 不高等问题, 特别是对于新兴的环境污染物以及待 开发的药物而言, 更是如此. 因此, 发展性能良好的 预测方法尤为重要. 3 pKa的预测 建立良好的pKa预测模型面临以下挑战: (1) 化合物构象灵活性, 有些化合物分子存在同分异构体或能够形成分子内氢键, 会显著影响其pKa值[39,40]; (2) 含有多个解离官能团, 因为要考虑micro-pKas, 使得pKa的预测变得更加复杂; (3) 可获得的用于建 立模型的pKa实验值. 尽管有些物质, 比如苯酚或羧 酸已有很多预测模型, 但是往往需要预测pKa的化合 物(新兴污染物或药物)不在现有模型的应用域内, 因此对于这些物质仍然需要精确的实验测定值. 在过去的20年间, 开发了大量的pKa预测模型, 涉及不同的计算水平、描述符类别以及统计分析方法 等. 当然, 回顾所有的研究方法不太现实, 因此本文试图总结大部分重要的研究成果, 列于表S2中[41~85]. 3.1 线性自由能关系(LFER)模型很多重要的pKa预测[41~45]都是基于线性自由能 关系, 使用Hammett方程, 认为取代基对pKa的贡献 是可以叠加的[42]:
其中, pKa0 是母体化合物的解离常数; 是常数, 与化合物的种类有关; i表示第i个取代基对母体化合 物解离常数的影响; m是取代基的个数. 该方法认为母体化合物携带了大部分化学信息, 取代基的贡献通过i来表征, 因此对于特定分子的 pKa预测受到如下限制: (1) 必须知道其母体化合物 的pKa值; (2) 必须已知所有取代基团的i 值. LFER模型是早期pKa预测研究中最常采用的方 法, 典型案例是Multi-CASE[44]. 共包含2464个有机 酸, 其中训练集1848个, 预测集616个, 也是最早使 用如此大规模数据的预测研究. 所有化合物按照中心解离官能团分成22个子集, 取代基(分子亚结构)对 中心官能团解离的影响通过碎片系数来表征, 例如, 对羧酸子集, 共定义了30个碎片系数. 此外, 还结合使用了分配系数、水溶解度、分子量、电负性、Hückel 分子轨道电荷密度以及硬度等分子结构参数. 基于训练集的模型对预测集的r2 =0.91, 均方根误差 RMSE=0.774; 基于全部数据的模型对214个药物分子的预测, r2 =0.70, RMSE=1.44[44]. 尽管是最早发展的方法, LFER模型目前仍用于商业软件或免费软件包, 例如EPIK[46,47]和SPARC[45]. SPARC结合LFER和扰动分子轨道理论, 将取代基对 pKa的影响分为共振效应、静电效应、溶剂化作用和氢键作用4个方面, 解决了键和电子离域对pKa的影 响. 这样处理使得SPARC可以计算micro-pKas, 并进而得到macro-pKas和其他相关性质, 例如滴定曲线等. 该方法应用于3685个化合物(包括含2~6个解离 中心的化合物), 总共4338个pKa值, 跨越30个数量级, 得到了很好的预测结果, RMSE为0.37. SPARC方 法最主要的应用局限在于其反应中心和取代基数据库有限, 有些性质还需要实验测定, 限制了对于新化合物的应用. 由此可见, LFERs预测方法成熟、性能稳定、应用范围广、计算速度较快, SPARC和ACD等软件都可以实现micro-pKas和macro-pKas的同时计算, 并具有良好的预测结果, 因此LFERs成为目前商业或免费软件包最常采用的计算方法. 然而, LFERs最主要的问题在于对新物质的预测应用受到其基础数据库所涵盖范围的限制. 如果新物质中含有数据库所不包含的分子碎片或者取代基, 常见的处理方法是使用已有的分子量最接近的同族原子或基团替代, 例如 SPARC没有对所有原子类型参数化, 实际应用中使用碳原子代替硅原子, 硫原子代替硒原子. 很显然, 这种处理会影响预测的准确度. 因此, 扩大数据集的涵盖范围、提高预测精度是以LFERs理论为主的预测方法的主要发展方向. 3.2 定量结构活性关系(QSPR)模型 pKa QSPR模型的基本假设是pKa值的大小由化 合物的分子结构决定, 而分子结构信息可以通过计算或实验测定而获得, 称之为分子描述符. 由于化合物结构和性质多样, 目前开发使用的描述符种类非 常多, 专著“Handbook of Molecular Descriptors”[86]列 出了1600余种不同的描述符, 使用这些描述符进而 通过统计学或者机器学习方法建立pKa预测模型. 预 测能力取决于模型所能够提取的描述符与pKa之间线 性或非线性相关关系的程度. 很多QSPR模型组合使 用了不同种类的描述符以及不同的统计分析方法, 具体详见表S2. 在众多小分子化合物pKa的QSPR模型中, 量子化学描述符因其具有清晰的物理化学意义、有助于进行机理解释而得到广泛应用. 由于计算量子化学描 述符要进行分子结构优化, 早期的、大数据集的研究 多使用半经验(semi-empirical)方法[39,49,50,65,87,88]. 随着计算机软硬件水平的发展, 计算能力不断提高, 从头算(ab inito)和密度泛函理论(DFT)也得到越来越多 的应用[55,57,58]. 超离域度和极化率是常使用的量子化学描述符 [39,40,50]. Tehan等 人 [39,50]使用这 2类描述符以及Coulson净原子电荷、最低未占据分子轨道能建立了417个有机酸(包括苯酚和羧酸, 并按解离基团类型、 能够形成分子内氢键以及芳环上取代基的位置分为6 个子集)和282个有机碱(包括苯胺、脂肪胺和杂环化 合物共9个子集)的pKa预测模型. 分子结构优化和频 率分析都是在半经验AM1水平上开展的. 对有机酸 各子集, r2 >0.80, RMSE |
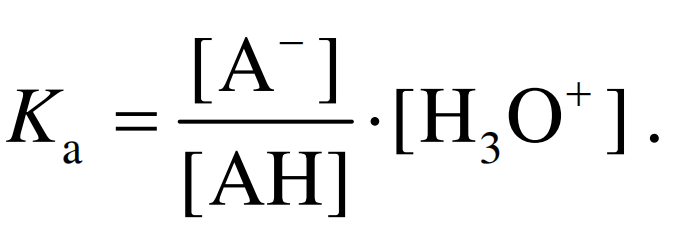 (1)
(1)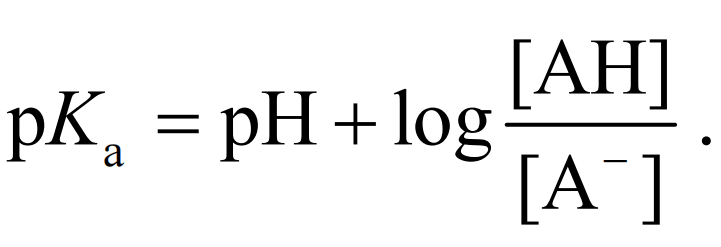 (2)
(2)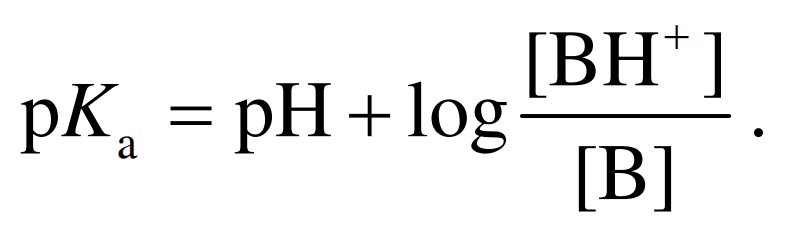 (3)
(3)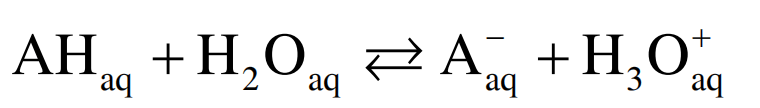
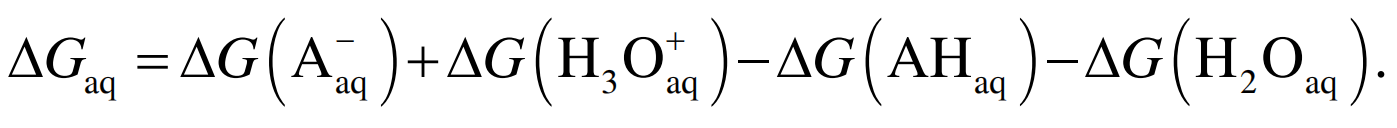 (4)
(4)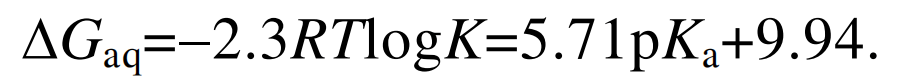 (5)
(5)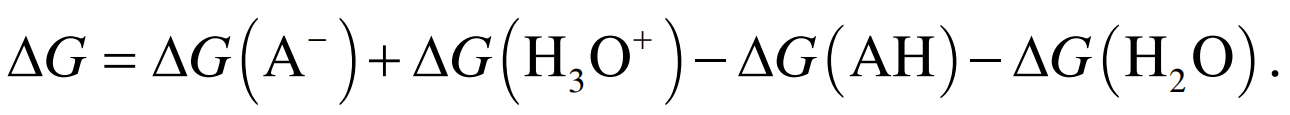 (6)
(6)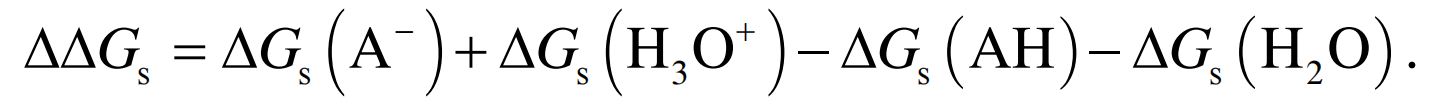 (7)
(7)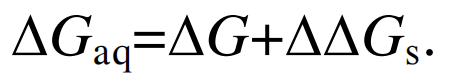 (8)
(8)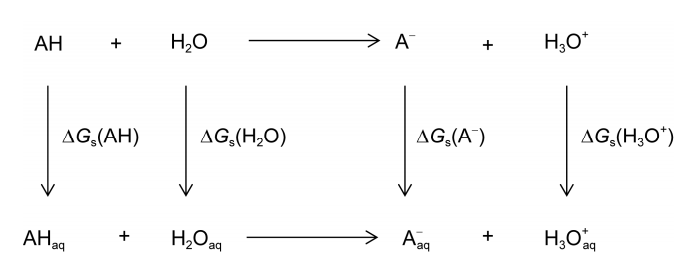 图1 气相和水相反应中质子从弱酸分子AH转移到水分子上的热动力学循环过程
图1 气相和水相反应中质子从弱酸分子AH转移到水分子上的热动力学循环过程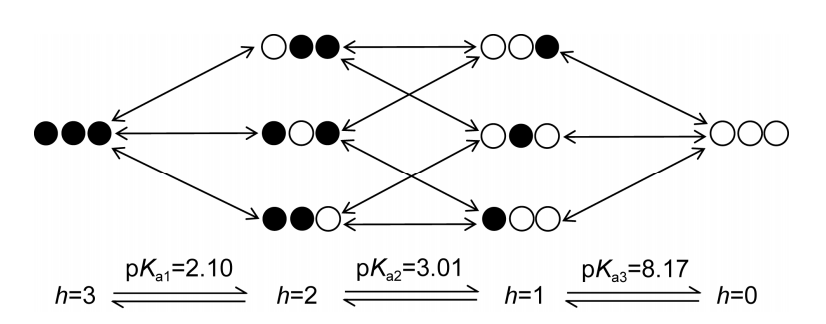
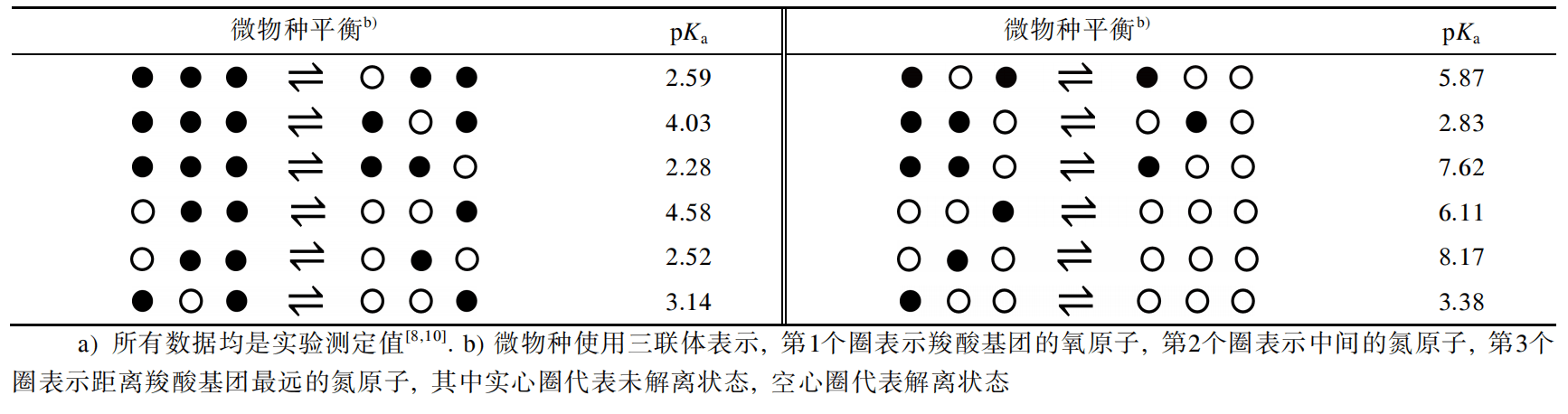 表 1 西替利嗪的 Micro-pKas
表 1 西替利嗪的 Micro-pKas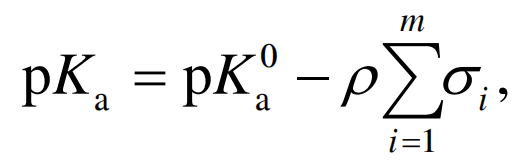
【本文地址】
今日新闻 |
推荐新闻 |