高变异药物生物等效性评价确切样本量计算 |
您所在的位置:网站首页 › phoenix研究缩写 › 高变异药物生物等效性评价确切样本量计算 |
高变异药物生物等效性评价确切样本量计算
|
影响BE研究样本量估计的因素包括几个方面:一是,进行2次单侧检验的显著性水准(α),一般设定为5%,对应于90%的置信区间(90%CI);二是,研究的把握度(power),系指两药事实上生物等效时,通过BE研究能正确地阐明生物等效的可能性;三是,两药物参数几何均数的比值(geometric mean ratio,GMR),如果真实比值GMR不为1,则需要增大样本量;四是,预期的个体内变异度;五是,研究设计类型。如重复的四周期交叉设计,每一个体接受试验药和参比药各2次,所需要的样本量就会比两周期的交叉设计减少一半。上述的因素确定后,最直接的影响因素就是监管部门规定的等效界值。 先考虑一般情况下BE研究的样本量估计事项。通常BE研究中生物等效性评价指标Cmax和AUC均按服从对数正态分布处理。因此,以下的所有考虑都是基于此假定进行的。先明确生物等效性推断的假设检验步骤。记试验药和参比药在对数尺度下的总体均数分别为
假设为平衡设计,在显著性水平α下,如果同时满足下列的2个条件,则可以拒绝无效假设,推断生物等效。
可见,进行生物等效性的推断需要在2个方向上同时进行2次单侧检验,因此该检验方法又称为双向单侧检验(two one-sided tests,TOST)。基于该统计推断原理,2次检验的α不变,但每次检验的β则和GMR直接相关,若GMR=1,两侧是对称的,但如果GMR不为1,则2次检验的β必然不同,分别记为下单侧检验的Ⅱ类错误概率(βL)和上单侧检验的Ⅱ类错误概率(βU)。就整个TOST推断而言,可理解为将总的β分解为βL和βU 2个部分。
根据统计学分布理论,研究表明,随机向量(Tlnθ1,Tlnθ2)服从自由度为n-2的双变量非中心t分布,所对应的非中心t分布参数分别为
具体的计算表达式如下。
其中,probt(·)为非中心分布的分布函数。由该式不难看出,如果设定α,给出期望达到的把握度,在GMR和个体内变异度(σw为个体内标准差)给定后,样本量是其中唯一的未知数。由该式难以直接求算出样本量,需要进行迭代运算。通常,迭代时先选定一个初始值n0(取整数),每次按步长1逐次迭代,直至求算的把握度大于并最接近于事先给定的把握度水平,此步对应的样本含量即为满足把握度要求的样本量。迭代初始值计算公式如下。
在实际的BE研究中,经常用变异系数(CV)表示个体内变异的情况。变异系数和标准差之间的互换关系式为
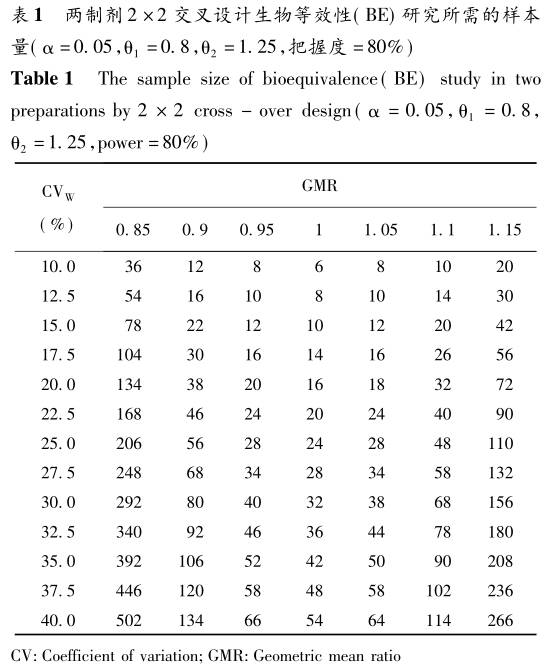
表1给出了把握度至少在80%,α为0.05,GMR为0.85到1.15,不同的个体内变异系数(CVw),几种不同设置下的总样本量。由于平衡的交叉设计需要偶数例数,计算出的奇数样本量都进行了舍入。
因为GMR和1/GMR为1两侧比值的对称点,理论上其样本量是完全相同的。故在同样CV和判断标准的情况下,GMR=1.1和GMR=0.9尽管在1的两侧差值对称,但比值并不对称,因此其样本量估计是不同的;而GMR=0.9与GMR=1.11在1两侧具有比值对称性,其样本量估计结果必然相同。 2 高变异药物BE研究确切样本量估计 对于高变异药物的BE研究,在FDA和EMA的相关指南中均采用了参比制剂校正的平均生物等效性(reference-scaled average bioequivalence,RSABE)评价方法。该方法利用参比制剂个体内变异对生物等效限值作标准化调整,其前提是对参比药进行重复测量设计。可采用部分重复三周期三序列交叉设计(TRR、RTR、RRT)和完全重复四周期两序列交叉设计(TRTR、RTRT)。 FDA指南中,RSABE方法对AUC和Cmax的等效性评价用
FDA还进一步说明了,当参比药个体内标准差σwr≥0.294(即CV=30%)时,可用RSABE评价方法。同时还要求BE研究的Cmax和AUC的GMR点估计必须在0.80~1.25内。该式表明了等效性评价的界值和参比药个体内标准差具有函数关系,进一步换算后的等效性评价的界值为 需要注意的是,当σwr=0.294(对应于CV=30%),所计算的界值为(77.0%,129.9%),与(80%,125%)并不重叠。 相比之下,EMA指南尽管也采用了RSABE评价方法,但其等效性评价的标准与FDA有明显的区别。当参比药的个体内CV大于或等于30%(Swr=0.294),用下式标准化后进行生物等效性评价,即
式中,Swr为参比药个体内标准差(相当于FDA指南中的σwr),EMA规定σω0=0.294。该式如果加以变换,则和FDA公式的形式几乎相同,只是σWR在数值上不同,即
如果Swr=0.294(对应于CV=30%),所计算的界值为(80%,125%),和非高变异药物BE研究的界值保持了连续性。进一步换算后的等效性评价的界值为 特别说明的是,EMA采用RSABE方法调整的等效范围最宽为69.84%~143.19%,对应的CV为50%,即当CV超过50%以后,无论变异系数如何变化,等效的限值不再放宽,统一使用(69.84%,143.19%)。FDA无此规定。 对于某一具体的高变异药物BE研究,一旦个体内变异系数或标准差(两者可随意互换)确定后,则可以求算出调整后的生物等效性限值,参照非高变异药物BE研究的样本量估计,可算得2×2交叉设计下的确切样本量。基于此,根据不同的设计类型,再推算出高变异药物BE研究的样本量,若采用部分重复的三周期交叉设计取其3/4,若采用完全重复四周期交叉设计取其1/2。高变异药物BE研究2×2交叉设计下确切样本量迭代计算的算式为
3 高变异药物BE研究确切样本量估计的SAS实现 在DATA步中用INPUT语句输入样本量估计的参数:alpha(I类错误概率水平α)、beta(Ⅱ类错误概率水平β,1-β为把握度)、CV(参比药个体内变异系数)、l(等效下限值)、u(等效上限值)、GMR(两药几何均数比值)。先计算RSABE方法调整的等效限值,继而计算样本量估计的初始值,然后采用DOUNTIL语句进行迭代运算直到power满足预先设定的水平。注意,对EMA指南要求,还需要采用IF语句控制CV大于50%后的等效限值固定不变。下面给出的是根据EMA指南要求的确切样本量计算SAS程序代码,可实现在不同的参数设定下算得相应的power和部分重复的三周期交叉设计和完全重复的四周期交叉设计的确切样本量。
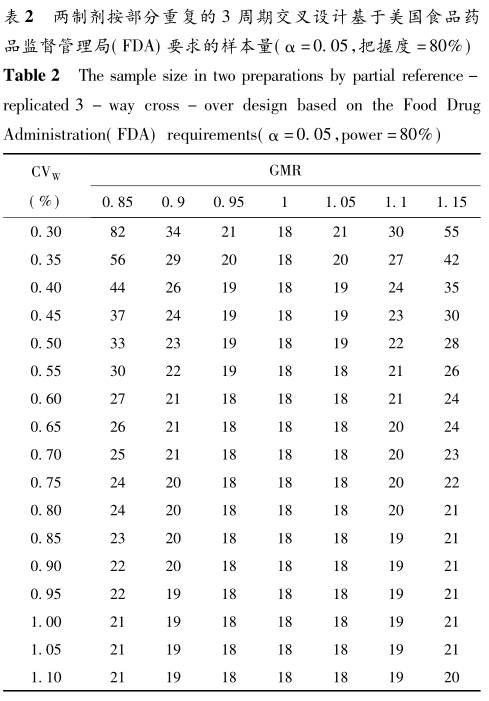
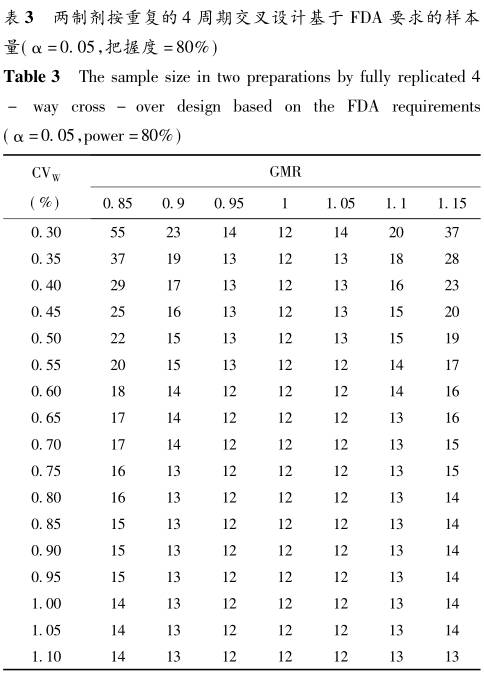
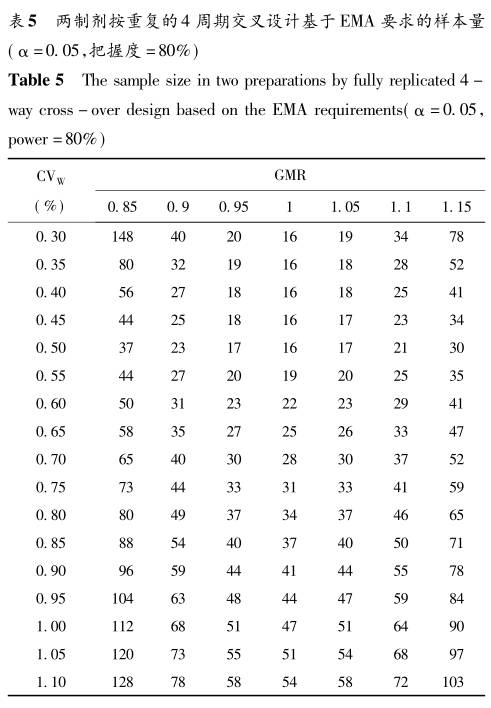
用本SAS程序代码,我们分别计算了两制剂不同重复类型交叉设计时,在不同的参数设定下,基于FDA指南和EMA指南要求的确切样本量,结果分别见表2~5。
4 讨论 本文基于高变异药物BE研究统计推断的原理和统计学分布理论,针对FDA和EMA对高变异药物BE研究的要求,给出三周期和四周期重复交叉设计的确切样本量估计结果。总的来看,在相同的样本量估计参数设置下,四周期交叉设计所需的样本量低于三周期交叉设计,FDA指南要求的样本量低于EMA要求。此外,FDA指南下的样本量,无论是三周期重复交叉设计还是四周期重复交叉设计,当GMR=1时,样本量在CV为30%~115%范围内保持最小且稳定不变(分别为18和12),而对GMR不为1的情形,呈现随CV增大样本量由大到小的变化趋势。而EMA指南下样本量在CV为30%~50%内与FDA指南呈现出相似的特征,但当CV超过50%时,所有GMR下的样本量均随CV增大而由小到大变化,只是GMR=1时的样本量仍保持最小。EMA指南下样本量的拐点出现在CV为50%处。由此可见,高变异药物BE研究的EMA指南对样本量的要求更高。 有学者曾利用公式近似法,基于FDA和EMA高变异药物BE研究指南要求,给出了不同参数设置下的样本量列表(Someswara Rao.K,2015)。将本文确切样本量估计方法与公式近似法计算的结果进行比较得出当GMR=1时,确切法的样本量均大于近似法,而对于GMR不为1的情形,确切法普遍小于或等于近似法。事实上,明确了高变异药物BE研究的方法学要求和相应的统计推断原理,进行样本量的统计学估计并不难。在实际应用中,完全可以借助专门的样本量估计软件进行求算。本课题组按照标准2×2交叉设计,借助PASS软件、nQuery软件等,在高变异药物的参数条件下进行了本文确切样本量方法与软件运算的比对,在表2~5范围内的样本量结果完全相同。 当然,任何研究的样本量估计绝不是从统计学上给出一个数字而已,有经验的统计专业人员往往会为临床研究者提供带有不同参数情景下的一组样本量结果,这样更有利于临床专业人员结合这些敏感性分析的结果,作出样本量的决策。高变异药物BE研究样本量的确定更是如此,不能完全依赖于统计学上计算的结果,还要对其他的一些因素,如临床研究机构资源等,进行综合考虑,这就需要另当别论了。 参考文献(略)返回搜狐,查看更多 |
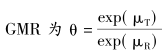

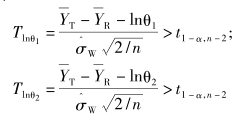

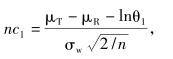
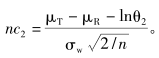
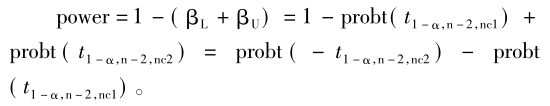
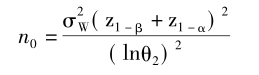
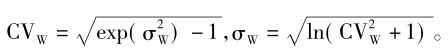
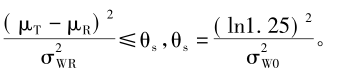

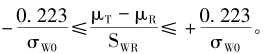
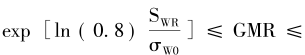
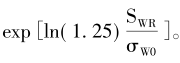
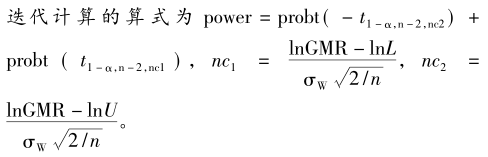





【本文地址】
今日新闻 |
推荐新闻 |