普通PCR、原位PCR、反向PCR和反转录PCR的 基本原理和操作步骤 |
您所在的位置:网站首页 › pcr的原理和操作 › 普通PCR、原位PCR、反向PCR和反转录PCR的 基本原理和操作步骤 |
普通PCR、原位PCR、反向PCR和反转录PCR的 基本原理和操作步骤
|
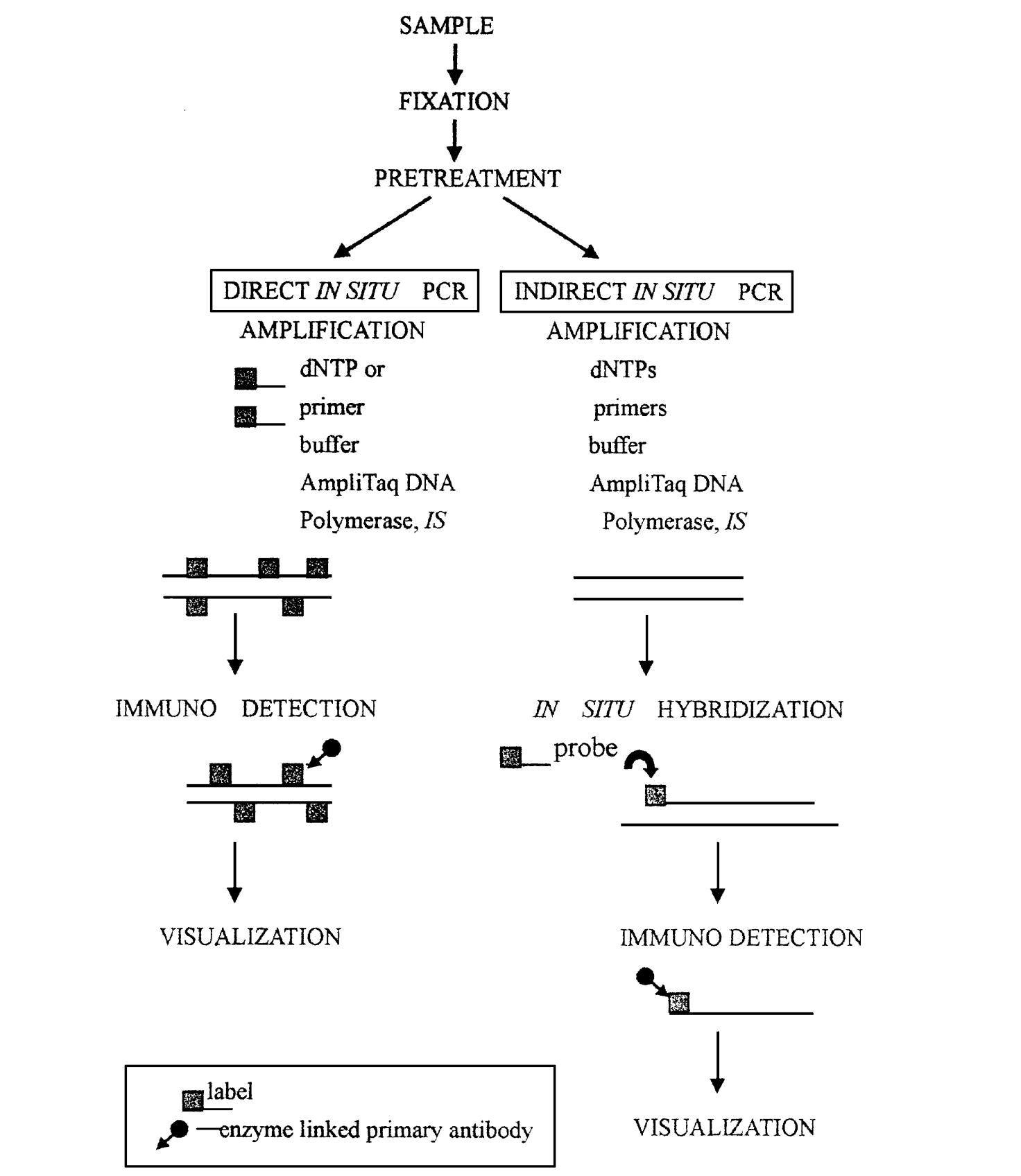
原位PCR 1 概述 自20世纪80年代中期PCR技术诞生以来, 研究人员就一直探索着将PCR技术与形态学研究结合起来。1990年, A.T. Haase等首创了原位PCR(In situ PCR, ISPCR), 当时称为 “细胞内PCR”。原位PCR是原位杂交和PCR结合的产物, 兼有二者的优点, 具有较好的灵敏性与专一性, 可同时获得组织、细胞及染色体的形态结构信息与分子信息, 又能检测出细胞中单拷贝或低拷贝的DNA、RNA序列。 2 原位PCR原理 原位PCR是通过在单细胞或组织切片上对特异DNA(或cDNA)进行PCR扩增, 然后采用原位杂交或免疫组织化学反应、荧光检测等技术进行细胞内特定核酸序列的检出及定位的分子技术。原位PCR技术的待检标本一般先经化学固定,以保持组织细胞的良好形态结构。细胞膜和核膜均具有一定的通透性,当进行PCR扩增时,各种成分,如引物,DNA聚合酶,核苷酸等均可进入细胞内或细胞核内,以固定在细胞内或细胞核内的RNA或DNA为模板,于原位进行扩增。扩增的产物一般分子较大,或互相交织,不易穿过细胞膜或在膜内外弥散,从而被保留在原位。这样原有的细胞内单拷贝或低拷贝的特定DNA或RNA序列在原位以呈指数极扩增,扩增的产物就很容易被原位杂交技术检查。 3 原位PCR类型 3.1 直接原位PCR 直接原位PCR是使用标记的引物或游离核苷酸进行原位PCR反应, 这种标记分子随后进入扩增产物中, 扩增结果可直接观察而不需要进行原位杂交(图1) 。 优点: (1)操作简便; (2)流程短; (3)省时。 缺点: (1)易发生引物错配或非特异性退火, 容易出现假阳性; (2)标记的引物会降低PCR效率。 3.2 间接原位PCR 间接原位PCR是在没有标记物的情况下进行PCR反应, 扩增反应结束后, 再用原位杂交技术来检测扩增的信号(图1)。 优点:可以克服由于DNA修复或引物错配引起的非特异性问题, 成为目前最广泛使用的方案。 缺点:需要进行扩增反应后的洗脱和原位杂交过程, 所需时间相对较长。
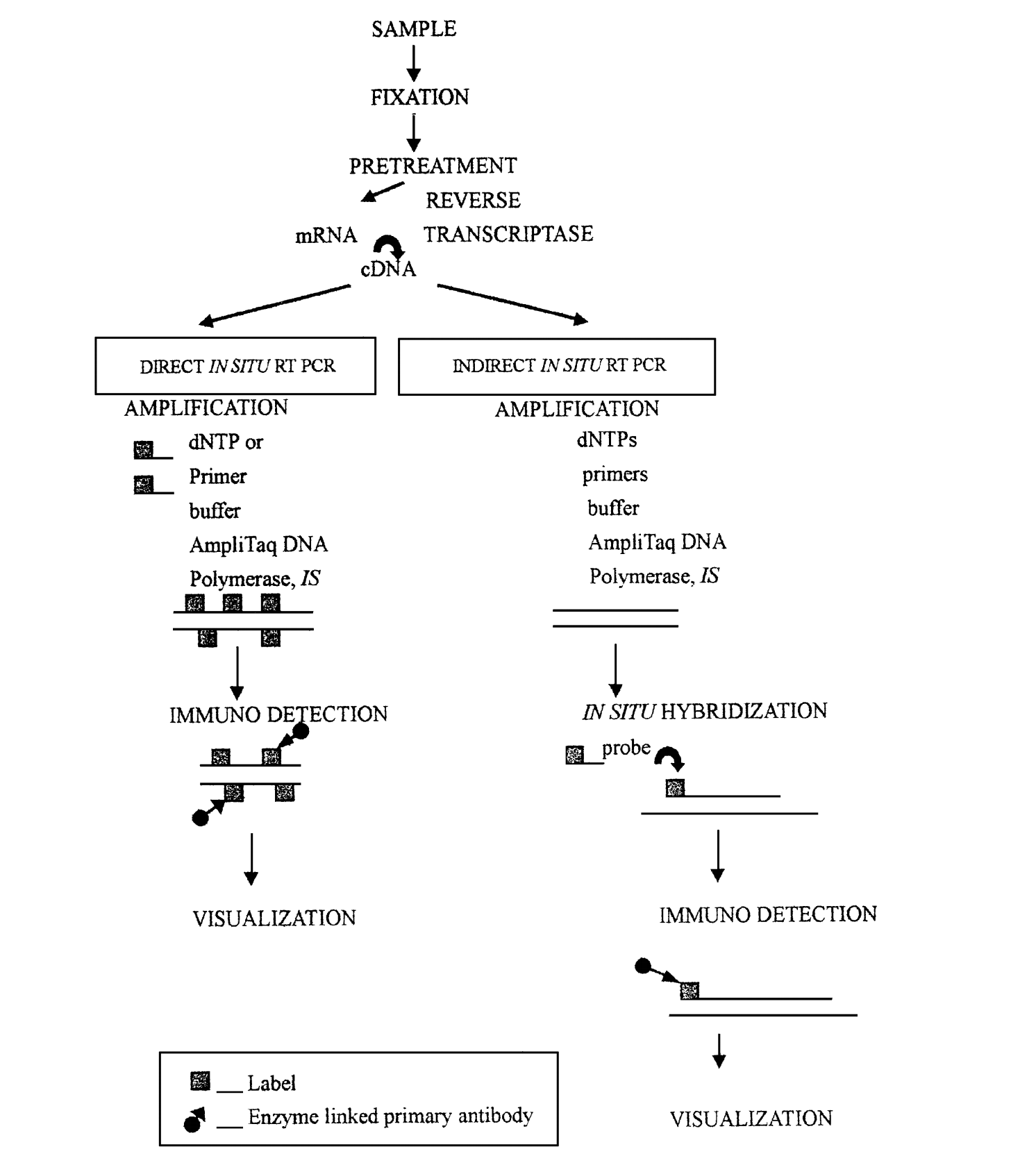
图1 直接原位PCR和间接原位PCR 3.3 原位反转录PCR 在原位 PCR 中加上一步反转录过程(RT) , 新形成的cDNA作为模板用于扩增, 这个过程叫做原位反转录PCR( in situ reverse transcription PCR, 简称原位RTPCR)。它又分为直接原位RTPCR和间接原位RTPCR两种(图2)。 原位RT PCR可用来检测细胞或组织中低拷贝的mRNA或特异基因的表达。 Tips:在原位 RTPCR 中, 首先要对组织样品进DNA 酶处理, 以破坏组织细胞中的DNA。
图2 原位反转录PCR 3.4 PCR原位再生式序列复制反应 Zebe等1994年率先提出了原位再生式复制反应( Selfsustained sequence replication reaction, 简称3SR反应)。它可作为原位RTPCR的一种选择方法用于完整的细胞和组织切片中低拷贝数mRNA的检测。 优点:有利于与免疫组织化学相结合。 4 植物原位 PCR 技术的操作步骤 植物原位 PCR 的基本步骤包括标本制备、预处理、原位扩增、后处理和检测等。 4.1 标本制备 固定剂的选择和固定的时间、温度因材料标本不同而各异。但固定条件的确定, 必须同时满足 2 个条件: ①保持染色体、细胞和组织标本形态结构的完整性; ②将原位程序中扩增效率的损失减到最小。 Tips: ①对于DNA的原位 PCR, 交联型固定剂(福尔马林、4%多聚甲醛、中性甲醛)较为适宜。交联型固定剂较利于蛋白质和核酸互相交联, 使扩增产物保留在原位,防止其向细胞外扩散或被洗脱。 ②固定时的温度也会对结果产生影响, 室温或 37℃固定24~48 h, 可获最强信号。 ③标本的年龄对实验结果影响也很大, 最好采用新鲜制备的不超过1个月的染色体标本。用新鲜固定的细胞做原位 PCR, 要比存档的石蜡切片做原位 PCR 敏感得多。 4.2 预处理 预处理可增加细胞的通透性, 促使反应试剂进入细胞内, 并使待扩增的靶序列暴露。在进行消化处理时, 酶种类的选择、浓度、温度和处理时间等条件的优化对于原位PCR都很重要。 Tips: ①胃蛋白酶、胰蛋白酶或蛋白酶K均可用于消化。推荐使用胃蛋白酶或胰蛋白酶, 因为蛋白酶K较难失活且易引起过度消化, 而胃蛋白酶、胰蛋白酶在以后的洗涤步骤中, 可通过提高pH抑制其活性, 或通过加热使其失活。 ②酶处理的方法取决于固定剂和标本类型。多聚甲醛和戊二醛固定的材料比FAA固定的材料需更长的酶处理时间, 且具小液泡的组织比具大液泡的组织预处理时间更长。 ③组织切片的细胞仍具有细胞壁, 应进行适宜的预处理。 ④对于解离好的染色体标本可不必进行蛋白酶消化, 而对于解离不充分的染色体标本要特别注意消化过度或消化不足。消化过度则扩增的靶序列易丢失, 消化不足则不利反应液的进入, 二者均会导致假阴性的出现。 ⑤有时候, 蛋白酶消化之前还需进行一些其他处理, 如进行蛋白酶消化之前, 进行了果胶酶处理。果胶酶处理可去除非特异性物质的污染。 4.3 原位扩增 原位PCR扩增体系中, 各组分的浓度和反应温度、时间、循环次数等的优化极为重要。由于玻片和细胞蛋白可能对反应液有所吸附, 因此, 与液相PCR相比, 原位PCR扩增溶液中各组分浓度相对较高。 Tips: ①液相PCR中最优的MgCl2浓度为1.5 mmol/L, 而原位PCR中为4.5 mmol/L; ②液相PCR中Taq DNA聚合酶的常规使用量为1.5 U, 而原位PCR要5 U才能产生较强的信号; ③反应温度因引物而定; ④循环次数不宜过高, 一般为20~30。 ⑤为了提高特异性, 可采用多步循环法或半套式扩增。 ⑥为了确定原位 PCR 检测的准确性, 必须设置阴性对照, 可设置为扩增反应液中不加入 Taq DNA聚合酶或引物。 ⑦间接法原位PCR反应液中的组分与普通PCR一样, 而直接原位PCR反应液中则包含了标记的引物或单核苷酸。在目前植物原位PCR的成功报道中, 都采用DIG-11-dUTP单核苷酸标记, 但dTTP与DIG-11-dUTP的使用浓度、二者的比例各不相 同。dTTP 和DIG-11-dUTP的终浓度比是原位扩增顺利进行的一个要素, 比值太高会使得DIG-11-dUTP在扩增过程中失去竞争力, 比值太低可能使信号扩散, DIG-11-dUTP 的浓度太高会形成空间阻碍。因此, 要优化出最适宜的浓度和比例。 ⑧反应液的体积应根据标本在玻片上的多少进行调整, 挑选合适大小的原位克隆环EasiSeal, 以防止反应液溢出或覆盖不足。 ⑨应根据不同植物材料、不同标本类型等各种因素, 优化出适宜的原位PCR体系,才能获得理想的结果。 4.4 后处理 大部分标本在原位扩增后要进行洗涤, 以降低背景和保留强阳性信号。针对不同的标本及其检测方法, 其后处理也有所不同。 Tips: ①为提高检测灵敏性和特异性, 使扩增产物保留在细胞内, 在洗涤完后, 用4%多聚甲醛或2%戊二醛对材料进行后固定, 可获得较好的效果。 ②对于染色体标本, 特别是纤维素酶和果胶酶处理较好的染色体标本, 其不似组织或细胞标本有细胞壁的保护, 扩增后的洗涤必须降低强度, 否则很容易使扩增信号丢失。 ③洗涤强度应根据检测信号进行优化, 若信号扩散可增加洗涤强度, 若出现假阴性, 则要降低洗涤强度。 4.5 检测 目前, 使用非放射性标记和检测DNA探针有生物素标记、地高辛标记和荧光素标记3 大检测系统。 Tips: ①采用地高辛标记检测系统, 用 NBT/BCIP(**四唑蓝/5-溴-4-氯-3-吲哚磷酸盐)显色进行检测。 ②采用 Anti-DIG-Fluorescein(荧光绿-地高辛抗体)进行免疫反应, 在显色反应中可选择DAPI (4,6-二脒基-2-苯基吲哚)或PI (碘化丙啶), 或DAPI/PI, 选择合适的荧光激发块进行检测。 ③原位PCR的检测系统可参考原位杂交的检测方法进行。 5 原位PCR的应用 5.1 在医学上的应用 (1)疾病中外源性基因检测; (2)诊断和某些相应基因的定位, 如HIV、HBV、EB 等病毒的检测。 5.2 在动物上的应用 主要用于流行病毒的检测。 5.3 在植物上的应用 (1)细胞或组织水平上的基因检测和定位; (2)染色体水平上的基因检测和定位; (3)单拷贝、低拷贝和多拷贝基因或特异序列的检测; ①检测植物材料是否受到外源致病基因的侵染和侵染部位; ②检测内源基因或特异序列在植物细胞或组织中的表达及分布; ③确定内源基因、特异序列及各种抗性基因在染色体上的位置; ④结合染色体带型分析, 构建物理图谱。 (4)杂种中亲本染色体的鉴定; (5)物种进化的研究; (6)遗传转化材料的分析。 虽然目前原位 PCR 技术在植物上的应用存在一定的困难, 但随着技术的成熟, 其应用前景必将广阔。只要在某种作物上建立了原位PCR体系, 则可为该类作物功能基因的研究奠定基础,在该类作物上的相关研究将会有更大的发展空间。 |
【本文地址】
今日新闻 |
推荐新闻 |