(22)基因功能注释和富集分析相关 |
您所在的位置:网站首页 › nr数据库是什么 › (22)基因功能注释和富集分析相关 |
(22)基因功能注释和富集分析相关
|
#基因结构预测 (拿到预测的蛋白序列、gff3文件那些) #基因功能注释##基因功能的注释依赖于上一步的基因结构预测,根据预测结果从基因组上提取翻译后的蛋白序列和主流的数据库进行比对,完成功能注释。 ##常用数据库: Nr:NCBI官方非冗余蛋白数据库,包括PDB, Swiss-Prot, PIR, PRF; 如果要用DNA序列,就是nt库 Pfam: 蛋白结构域注释的分类系统 Swiss-Prot: 高质量的蛋白数据库,蛋白序列得到实验的验证 KEGG: 代谢通路注释数据库. GO: 基因本体论注释数据库 BLASTP检索基本流程:下载数据库,构建BLASTP索引(想要用谁检索,就对谁构建索引,索引是为了方便快速检索的),数据库检索,结果整理。其中结果整理需要根据BLASTP的输出格式调整。 都是先以南极石耳为例跑一下流程,后期如果批量做的话还需要适当改一下代码~ 补充疑惑: 1)为啥要进行富集分析,核心逻辑是什么? 就是说,咱找到了一系列的基因,咱想知道这些基因有没有一个统一的功能、通路等等,所以通过数据库将其富集起来,看看XX占的最多,作用最大 2)准备材料是什么呢? 一般都是用预测到的蛋白文件,需要有目的基因,有想研究的对象(比如,与寒冷相关基因,正选择基因、快速进化基因等等) 1. NR 注释 1.1下载真菌的refseq数据库第一次用下载就行了,以后就可以直接用,不用重复下载~ mkdir -p ~/Qxy/ncbi-blast-qxy/db/ cd ~/Qxy/ncbi-blast-qxy/db/ # download 下载了NCBI里Refseq数据库中的Fungi数据 wget -4 -nd -np -r 1 -A *.faa.gz ftp://ftp.ncbi.nlm.nih.gov/refseq/release/fungi/ #服务器下载格式有误,解压失败,本地下载再上传 mkdir -p ~/Qxy/ncbi-blast-qxy/RefSeq zcat *.gz > ~/Qxy/ncbi-blast-qxy/RefSeq/fungi.protein.faa # build index 对该数据库fungi建立索引 ~/anaconda3/bin/makeblastdb -in fungi.protein.faa -dbtype prot -parse_seqids -title RefSeq_fungi -out fungi 1.2正式分析 cd ~/Qxy/qxycexu/U.antarctica/funannotate mkdir Nr cd Nr ln -s /ifs1/User/wuqi/Qxy/knowngenome/orthofinder/Umbilicaria_antarctica.proteins.fasta ./ # search 将自己的数据拿去与nr库进行比对 nohup diamond blastp --db ~/Qxy/ncbi-blast-qxy/RefSeq/fungi.protein.faa --query Umbilicaria_antarctica.proteins.fasta --out Nr.xml --outfmt 5 --sensitive --max-target-seqs 20 --evalue 1e-5 --id 10 --index-chunks 1 & # 比对后得到.xml文件,利用下列脚本进行处理 ~/Qxy/qxyjiaoben/parsing_blast_result.pl --out-hit-confidence --suject-annotation Nr.xml > Nr.tab ~/Qxy/qxyjiaoben/nr_species_distribution.pl Nr.tab > Nr_species_distribution.txt ~/Qxy/qxyjiaoben/gene_annotation_from_Nr.pl Nr.tab > Nr.txt ###研究某个基因!!!将基因与基因组进行比对 mkdir -p ~/13.functional_annotation/00.blast_to_genome cd ~/13.functional_annotation/00.blast_to_genome # 构建蛋白质数据库 makeblastdb -in ~/00.incipient_data/data_for_gene_prediction_and_RNA-seq/Malassezia_sympodialis_V01.protein.fasta -dbtype prot -title Malassezia_sympodialis_V01.protein -parse_seqids -out Malassezia_sympodialis_V01.protein -logfile protein.log # 构建核酸数据库 makeblastdb -in ~/00.incipient_data/data_for_genome_assembling/assemblies_of_Malassezia_sympodialis/Malassezia_sympodialis.genome_V01.fasta -dbtype nucl -title Malassezia_sympodialis_V01.genome -parse_seqids -out Malassezia_sympodialis_V01.genome -logfile Malassezia_sympodialis_V01.genome.log # 在https://www.ncbi.nlm.nih.gov/gene/数据库搜索"3-phytase a",点击Send to,选择File,在Format中选择UI List,下载文件保存为gi.ncbi_gene_phyA.list #一个是对核酸进行比对 cp ~/00.incipient_data/data_for_functional_annotation/gi.ncbi_gene_phyA.list ./ ncbi_acc_or_gi_to_fasta.pl gi.ncbi_gene_phyA.list 10 > phyA.nucl.fasta perl -p -e 's/>(\S+).*/>$1/; s/\|/_/g' ~/00.incipient_data/data_for_functional_annotation/phyA.nucl.fasta > phyA.nucl.fasta blastn -query phyA.nucl.fasta -db Malassezia_sympodialis_V01.genome -out blastn.out -evalue 1e-5 -outfmt 7 -num_threads 4 # 在https://www.uniprot.org/uniprot/搜索"3-phytase a" AND reviewed:yes,并下载数据 #一个是对蛋白进行比对 perl -p -e 's/>(\S+).*/>$1/; s/\|/_/g' ~/00.incipient_data/data_for_functional_annotation/uniprot-_3-phytase+a_-filtered-reviewed_yes.fasta > phyA.pep.fasta blastp -query phyA.pep.fasta -db Malassezia_sympodialis_V01.protein -out blastp.out -evalue 1e-5 -outfmt 7 -num_threads 4注:这个我用本地blast就可以做到~(之前教程里面有写) 2.进行 Swiss-Prot 注释 2.1下载数据库 mkdir -p ~/Qxy/Swiss-Prot cd ~/Qxy/Swiss-Prot # download 下载UniProtKB/Swiss-Prot数据库中的蛋白质文件 wget -4 -q ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete/uniprot_sprot.fasta.gz gzip -d uniprot_sprot.fasta.gz # builid index 构建索引 ~/anaconda3/bin/makeblastdb -in uniprot_sprot.fasta -dbtype prot -title swiss_prot -parse_seqids 2.2正式分析 cd ~/Qxy/qxycexu/U.antarctica/funannotate mkdir Swiss-Prot cd Swiss-Prot ln -s /ifs1/User/wuqi/Qxy/knowngenome/orthofinder/Umbilicaria_antarctica.proteins.fasta ./ #使用添加了名字的蛋白序列去做 # search 运行比对,挺快的,结果是一个.xml文件 diamond blastp --query Umbilicaria_antarctica.proteins.fasta --db ~/Qxy/Swiss-Prot/uniprot_sprot.fasta --out uniprot_sprot.xml --outfmt 5 --sensitive --max-target-seqs 20 --evalue 1e-5 --id 10 --index-chunks 1 # 运用脚本整合结果,速度挺快的,得到.tab和.txt文件 ~/Qxy/qxyjiaoben/parsing_blast_result.pl --out-hit-confidence --suject-annotation uniprot_sprot.xml > uniprot_sprot.tab ~/Qxy/qxyjiaoben/gene_annotation_from_SwissProt.pl uniprot_sprot.tab > SwissProt.txt 2.3结果展示最终得到.xml,.tab和.txt结果文件,.xml是最原始的结果,文件大,内容全,但是不方便阅读 .tab显示了每个蛋白比对到了谁,比对上多少,P值多少等等
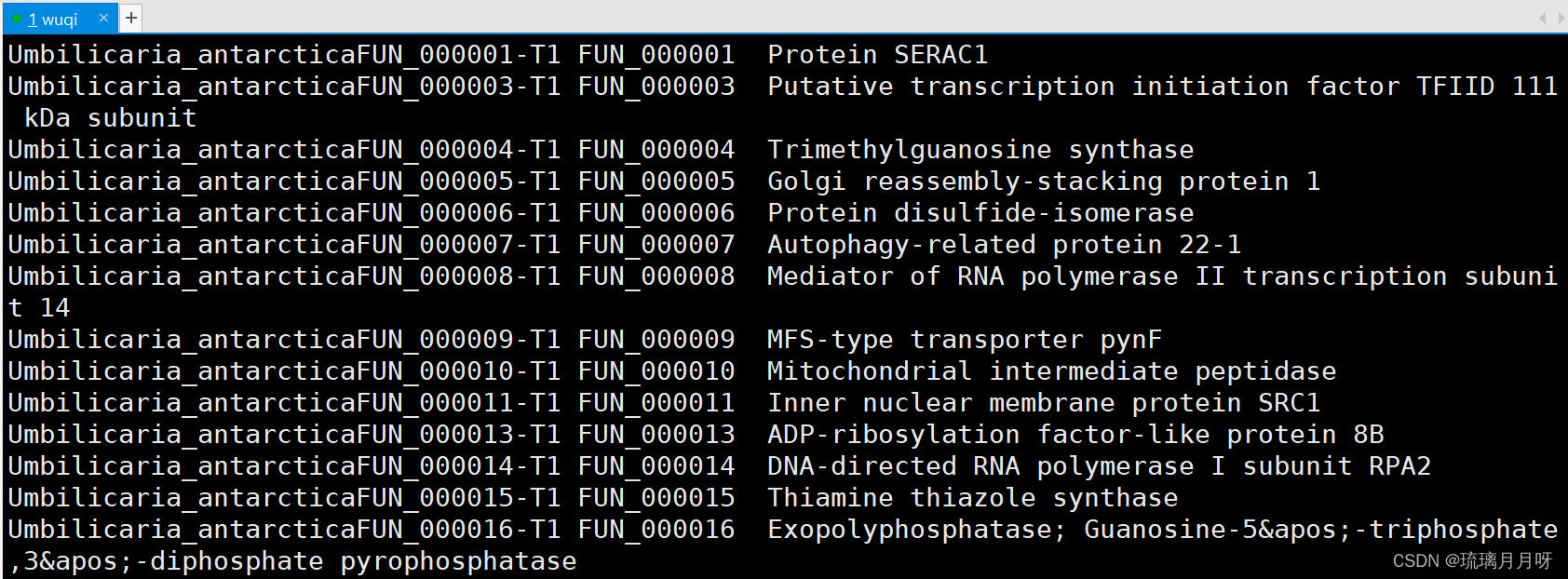
.txt比较容易阅读,显示蛋白注释到的功能
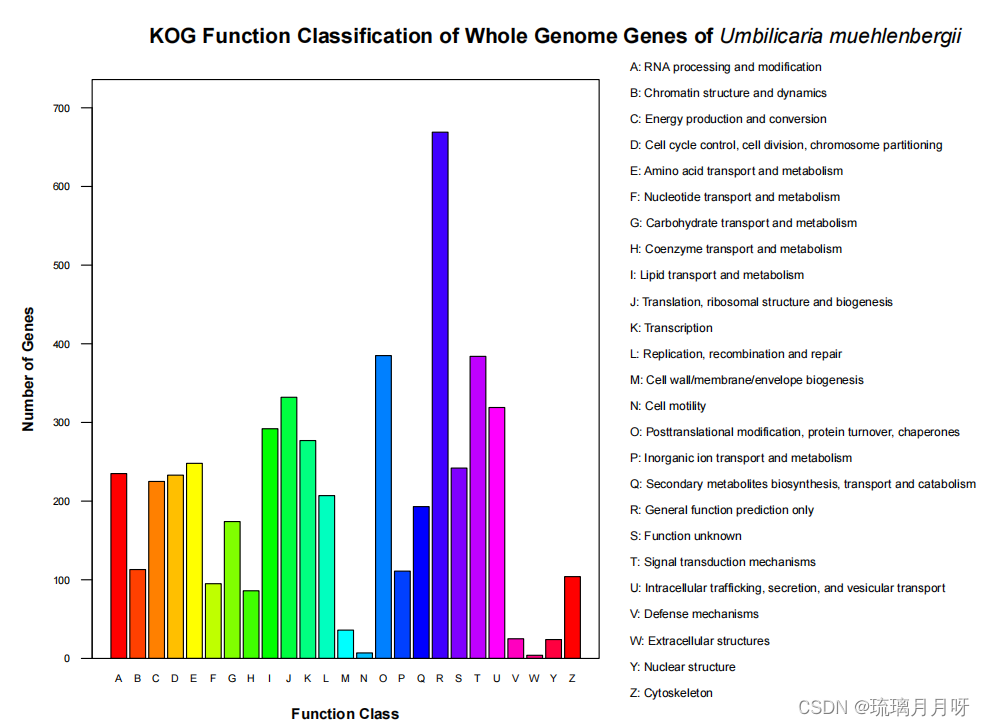
KOG注释分为25个类别A-Z(没有X),结果文件是kog.xml,经过处理得到out.tab、out.annot、out.class out.tab表格展示的是XX比对到XX,以及匹配开始结束、P值那些,比较详细 out.annot表格更加直观一些,就是XX注释到了XX,包含KOG编号 out.class展示的是这25大类分别包含哪些基因,且对应的KOG是谁 然后又汇总形成了KOG.txt,更具体,并进行作图
注:这是对整个基因组进行了KOG注释,然后作图,意思就是在这整个基因组中,主要分了25大类,每一大类又分别含有哪些基因
这25大类注释依次是 A: RNA processing and modification RNA加工和修饰 B: Chromatin structure and dynamics 染色质结构和动力学 C: Energy production and conversion 能源生产和转换 D: Cell cycle control, cell division, chromosome partitioning 细胞周期调控,细胞分裂,染色体分裂 E: Amino acid transport and metabolism 氨基酸运输和代谢 F: Nucleotide transport and metabolism 核苷酸转运和代谢 G: Carbohydrate transport and metabolism 碳水化合物运输和代谢 H: Coenzyme transport and metabolism 辅酶运输和代谢 I: Lipid transport and metabolism 脂质转运和代谢 J: Translation, ribosomal structure and biogenesis 翻译、核糖体结构和生物发生 K: Transcription 转录 L: Replication, recombination and repair 复制、重组和修复 M: Cell wall/membrane/envelope biogenesis 细胞壁/膜/包膜生物发生 N: Cell motility 细胞运动 O: Posttranslational modification, protein turnover, chaperones 翻译后修饰,蛋白质周转,伴侣 P: Inorganic ion transport and metabolism 无机离子运输和代谢 Q: Secondary metabolites biosynthesis, transport and catabolism 次生代谢物的生物合成、运输和分解代谢 R: General function prediction only 一般功能预测 S: Function unknown 功能未知 T: Signal transduction mechanisms 信号转导机制 U: Intracellular trafficking, secretion, and vesicular transport 细胞内运输、分泌和囊泡运输 V: Defense mechanisms 防御机制 W: Extracellular structures 细胞外结构 Y: Nuclear structure 核结构 Z: Cytoskeleton 细胞骨架 4.进行eggNOG 注释零基础快速完成基因功能注释 / GO / KEGG / PFAM... - 简书 生信石头的教程
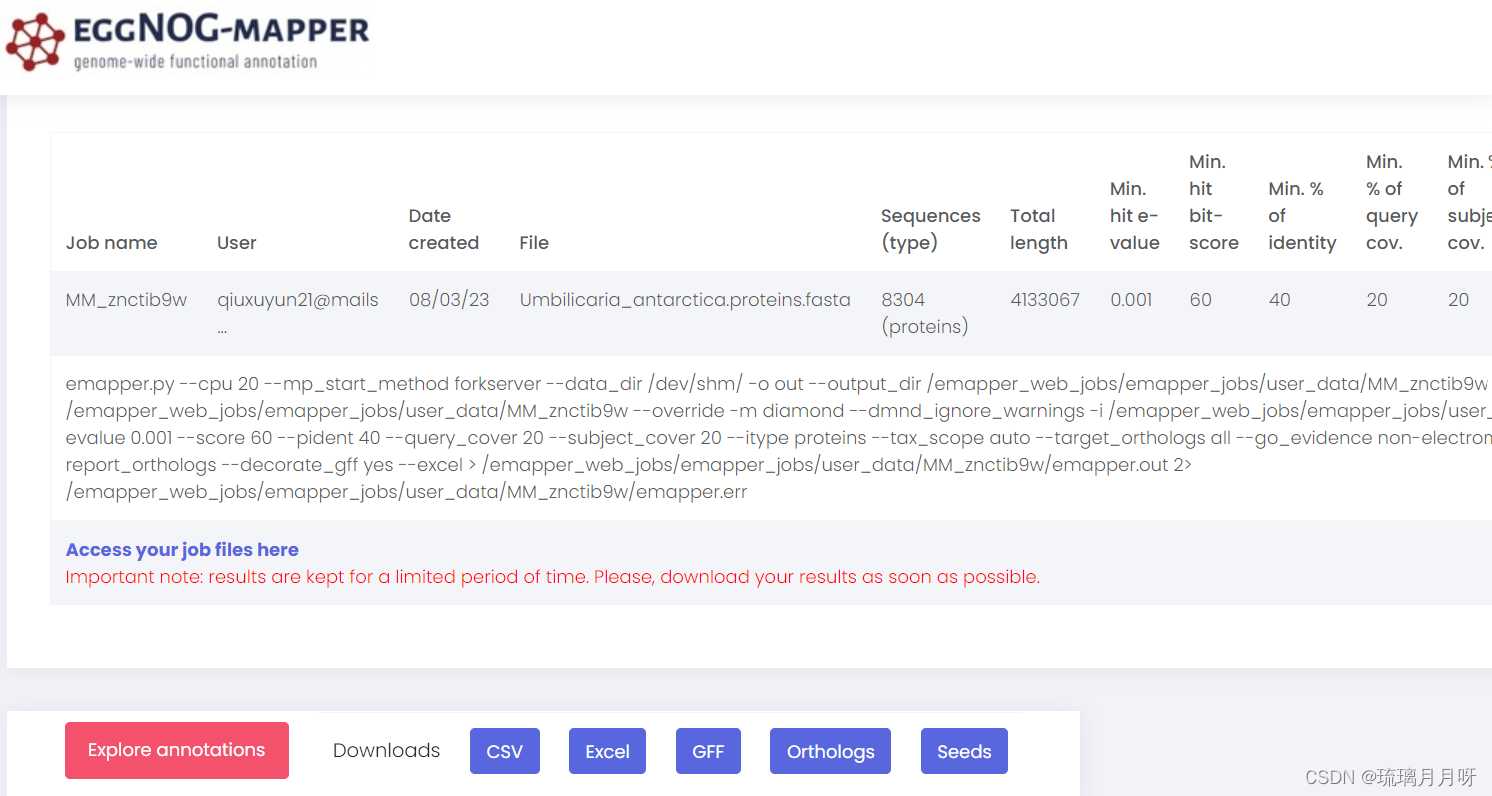
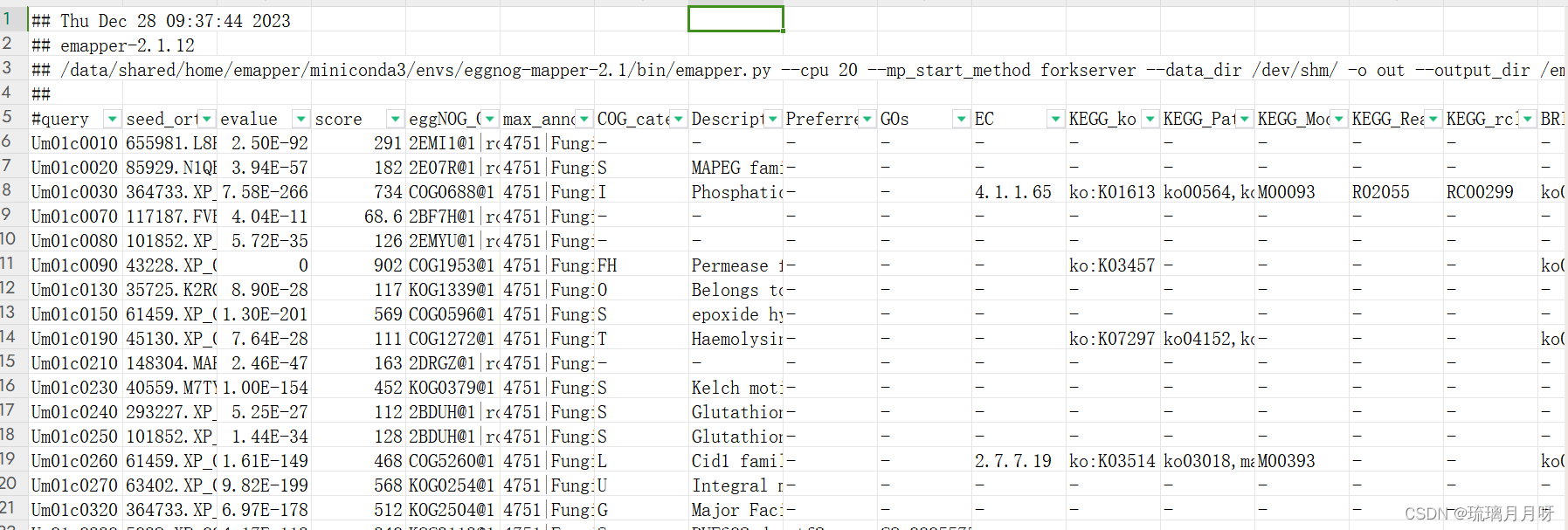
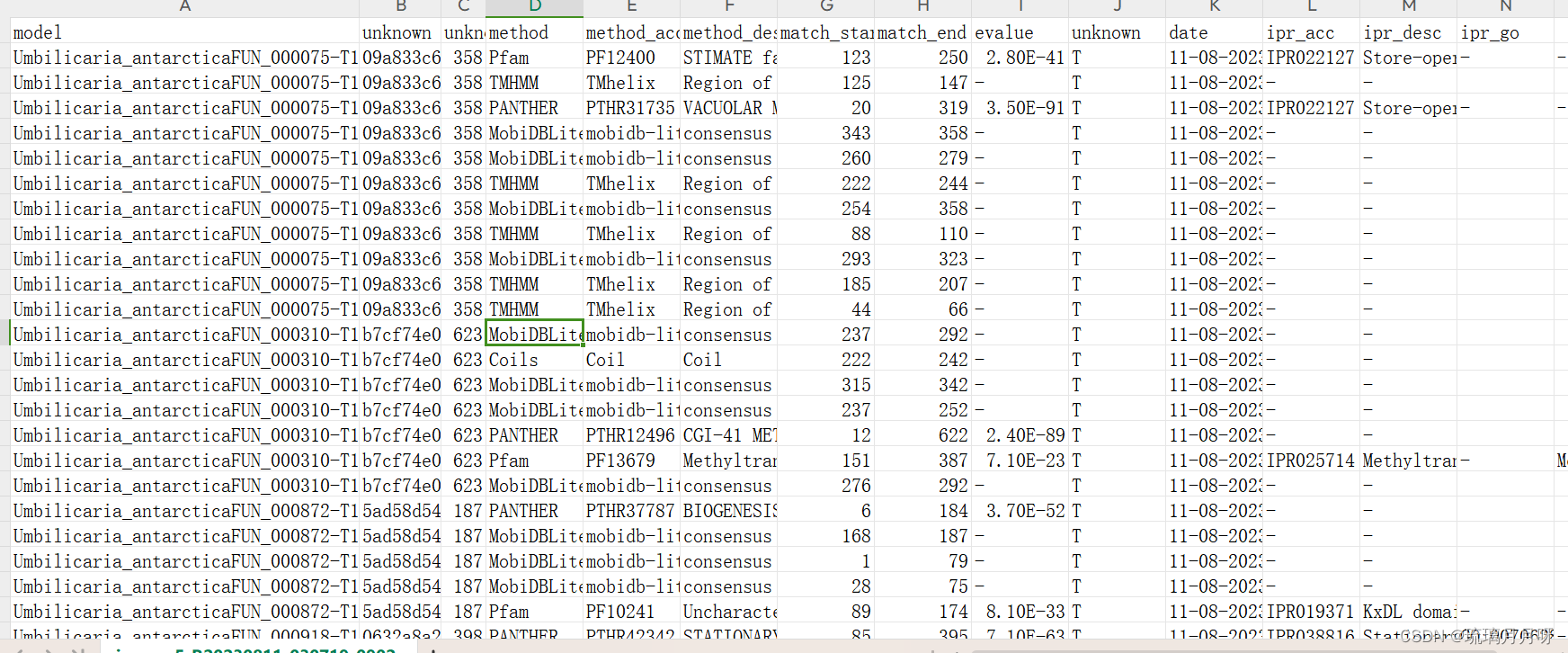
!!!eggNOG官网的网页工具提交进行注释http://eggnog-mapper.embl.de/提交蛋白序列 #注,蛋白序列用跑orthofinder的那些,每条都有物种名,上传序列、填写邮箱、!有一个邮件,点进去然后开始运行!,速度还行 start !!!要点击开始运行啊!!!(注:这次结果文件是自己选择download不同格式,我下载了xlsx格式,然后不用上传服务器了,直接本地操作就行) 运行结束之后会再发一个邮件,然后点击链接进去就行了,或者也可以隔一段时间看看网页是不是状态变成了done #目标结果文件是out.emapper.annotations #grep -v -P "^#" out.emapper.annotations | cut -f 1,8 > eggNOG.txt #这是跟着clf取了第1和第8列,知道基因和对应功能 我这次eggNOG结果文件是这样的 1)query:输入序列的名称。 2)seed_ortholog:匹配到的种子序列的注释信息。 3)evalue:输入序列与种子序列的匹配E-value阈值。 4)score:输入序列与种子序列的匹配得分。 5)eggNOG_OGs:与输入序列匹配的EggNOG orthologous groups(OGs)。 6)max_annot_lvl:在输入序列的注释中提供的最大注释级别。 7)COG_category:Clusters of Orthologous Groups(COG)分类。 8)Description:该序列的功能描述。 9)Preferred_name:该序列的首选或标准名称。 10)GOs:Gene Ontology(GO)注释信息。 11)EC:对应的酶学注释号。 12)KEGG_ko:对应的KEGG Orthology(KO)号。 13)KEGG_Pathway:KEGG通路信息。 14)KEGG_Module:KEGG模块信息。 15)KEGG_Reaction:KEGG反应信息。 16)KEGG_rclass:KEGG反应分类信息。 17)BRITE:BRITE功能层次结构注释。 18)KEGG_TC:KEGG传输物质分类信息。 19)CAZy:碳水化合物活性酶家族注释信息。 20)BiGG_Reaction:输入序列与BiGG数据库中的反应匹配的信息。 21)PFAMs:序列中与PFAM数据库匹配的信息。这个应该是最新的版本,然后第一列、第10列GO号、第12列KO号比较重要,其他的后续分析可能会用到 (自己需要什么自己直接在EXCEL中提取就行了,别再上传服务器再写代码啥的,你又写不明白) 东西多着呢,一个eggNOG基本上把所有东西都注释出来了! 5.进行Interpro 注释 5.1在线网站操作进入Interpro网站 http://www.ebi.ac.uk/interpro/search/sequence/ 但是该网站有个bug,一次只能提交25条序列,so用TBtools进行批量操作,粘贴序列,选择浏览器,设置输出目录,然后start就行 (!!!但是上面在线操作结果无法下载!!!) 5.2服务器操作 #服务器操作 mkdir interpro cd interpro ln -s /ifs1/User/wuqi/Qxy/knowngenome/orthofinder/Umbilicaria_antarctica.proteins.fasta ./ # wget https://raw.githubusercontent.com/ebi-wp/webservice-clients/master/python/iprscan5.py #在网上下载iprscan5.py脚本,然后还给服务器的python补充了个xmltramp2模块,然后就可以在服务器批量运行了 sed 's/\*//g' Umbilicaria_antarctica.proteins.fasta > cleanseq.fa #先去除序列中的XX符号,but好像去了没去一个样 nohup python3 ~/Qxy/interpro5/iprscan5.py --multifasta Umbilicaria_antarctica.proteins.fasta --maxJobs 25 --useSeqId --email [email protected] --outformat tsv & !!!要一直保持网络连接!!!一旦断网就不行了~ 然后将cleanseq.fa文件传输到本地,利用TBtools中merge and split功能,传输XX.fa文件,选择输出文件前缀(输出目录在E盘TBtools文件夹下),选择Record for files,输入1000,则按照1000条序列来对原文件进行分割 nohup python3 ~/Qxy/interpro5/iprscan5.py --sequence output.0.split.fa --stype p --email [email protected] --outformat tsv & 5.3结果解读结果文件是.tsv,导到本地是这样,大概是将蛋白序列按照结构域或基因家族分类,不同库注释了一下 Interpro是集成了蛋白质家族、结构域和功能位点的非冗余蛋白质特征序列数据库, Interpro数据库成员包括Coils 、Gene3D、Pfam、PRINTS、ProSitePatterns、 ProSiteProfiles、 SMART、 SUPERFAMILY、 TIGRFAM、 ProDom、 PIR 数据库。采用 interproscan 软件可以对新蛋白质序列通过序列比对或者 HMM 算法等搜索与 interpro 蛋白质特征序列匹配预测蛋白质各种结构功能域、信号肽、跨膜特征、蛋白质螺旋结构等 这是加了一下首行标题的,自己参考别人注释文件加的
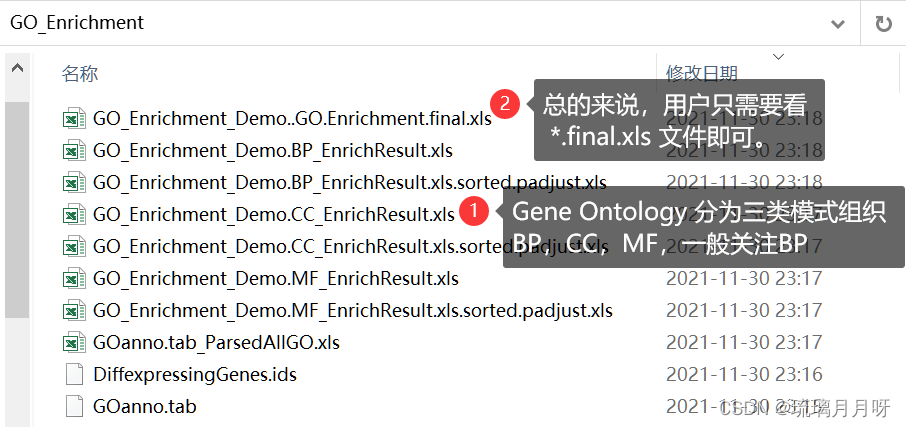
6.2GO分类 #输入go-basic.obo、go.annot、goslim_agr.obo,但是最后一个文件没找到 6.3GO富集分析 #需要有目的基因,上调or下调 (这是教材里的,做不下去了) 6.3.1用TBtools进行GO富集分析「GO富集分析」从原理到实践 ~ 零基础掌握-CSDN博客 直接看原文,我的输入数据是根据eggNOG调整来的 GO-Slim这里选一下yeast,然后其他的根据教程输入就行
会得到一系列结果文件,然后不知道是什么bug,反正要自己手动将.final.xls文件补充一下 然后继续用TBtools进行可视化
补充: GO的结果展示可以是柱状图,也可以是有向无环图
这就是有向无环图,可以用TopGO做,但是我做出来结果不太理想,一片红或者是最后有0
KAAS网址:http://www.genome.jp/kaas-bin/kaas_main Search program选择GHOSTX(快速且准确率高) Quern sequences选择上传文件(还是用跑orthofinder的蛋白文件) Query name命名这次注释工作 Email填写邮箱地址 GENES data set选择对应的下面这些 ani, afm, nfi, aor, ang, afv, pcs, pdp, cim, cpw, ure, pbl, abe, sce, ago, kla, kmx, lth, vpo, zro, cgr, ncs, tpf, ppa, dha, pic, pgu, spaa, lel, cal, yli, clu, clus, caur, slb, pkz Assignment method选择BBH(双向匹配) 然后点击compute开始计算,然后会说Job Request,去邮箱里点击submit那个的链接进行提交,然后会得到Result结果链接 7.2富集分析需要准备KAAS注释文件、KEGG信号通路以及目标基因文件 7.2.1用TBtools进行富集分析TBtools 实用教程 - 点点点,KEGG Pathway 通路富集分析 - 《TBtools Cookbook - TBtools 帮助手册教程》 - 极客文档 需要一个背景文件Fungi.20211128.TBtoolsKeggBackEnd 需要选择真菌Fungi 得到结果文件
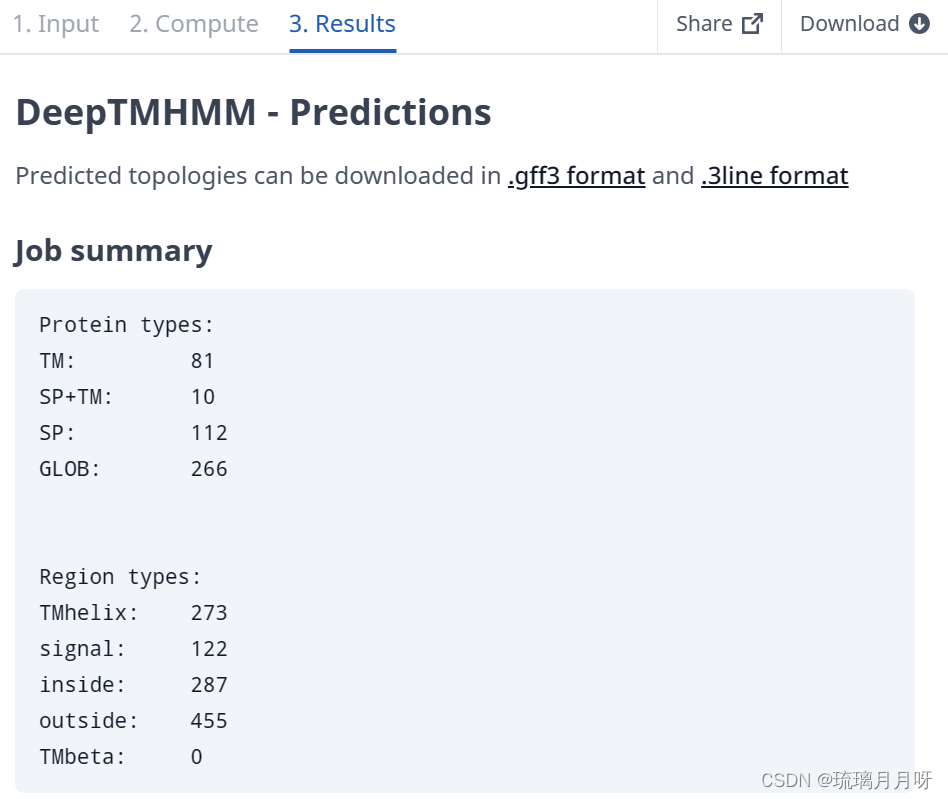
也可以进行可视化 好像我的.final.xls文件有点问题,不如直接用不带后缀的文件 8.CAZY annotation使用在线网站进行注释dbCAN3网址:https://bcb.unl.edu/dbCAN2/(1和2都已经过时了) 点击Annoation进入注释页面,然后输入邮箱,选择比对模式(前三),输入序列,然后提交submit 好像前面eggNOG已经把CAZY给注释出来了 补充:碳水化合物活性酶(Carbohydrate-Active enzymes, CAZy)是一大类很重要的酶,分为糖苷水解酶类、糖基转移酶类、多糖裂解酶类以及糖酯酶类,具有降解、修饰及生成糖苷键的功能。(催化作用) 结果解读,得到XX.xlsx表,XX注释相关总表,XX.seq序列相关表 凡是在XX.xlsx表里的,都是被注释到是CAZY的,在表里可以看到这个网站分别用了HMMER、DIAMOND、dbCAN_sub三种方式进行比对,并给出相应结果,后面是信号肽,以及会统计得到了几种工具的结果,第2列EC那个,表示酶的名字 XX.seq是它对应的序列 9.Pfam注释 #para_hmmscan.pl --outformat --cpu 4 --hmm_db /opt/biosoft/hmmer-3.3.1/Pfam-A.hmm ../proteins.fasta > Pfam.tab 9.1下载安装Pfam数据库 wget ftp://ftp.ebi.ac.uk/pub/databases/Pfam/releases/Pfam33.1/Pfam-A.hmm.gz gunzip -d Pfam-A.hmm.gz hmmpress Pfam-A.hmm 9.2使用hmmscan进行Pfam注释 cd ~/Qxy/qxycexu/U.antarctica/funannotate mkdir Pfam cd Pfam ln -s /ifs1/User/wuqi/Qxy/knowngenome/orthofinder/Umbilicaria_antarctica.proteins.fasta ./ ~/Qxy/qxyjiaoben/para_hmmscan.pl --outformat --cpu 4 --hmm_db ~/Qxy/Pfam/Pfam-A.hmm Umbilicaria_antarctica.proteins.fasta > Pfam.tab cut -f 1,2,7 Pfam.tab | perl -e '; print ' | ~/Qxy/qxyjiaoben/gene_annotation_from_table.pl - > Pfam.txt这个前面eggNOG也注释了,但是好像有点不太一样,这个更具体一些 10.真菌的 Transcription Fatcor 注释(需要interpro的结果文件) cd ~/Qxy/qxycexu/U.antarctica/funannotate mkdir TF cd TF interpro2tf_for_Fungi.pl ../functional_annotation.InterPro.tab --out_prefix TF 11.分泌蛋白注释这是一个层级筛选过程,分泌蛋白都有信号肽,so先分析信号肽;再分析跨膜区,要是有跨膜区则会被固定到膜上,不会成为分泌蛋白,所以要选择没有跨膜区的;再分析GPI锚定位点,选择没有的,不然也会被固定在膜上,无法分泌出去;最后进行亚细胞定位,选取定位到胞外的蛋白作分泌蛋白基因,得到最终结果。 11.1信号肽注释,分泌蛋白都具有信号肽 cd ~/Qxy/qxycexu/U.antarctica/funannotate mkdir secreted_protein cd secreted_protein mkdir singalp cd singalp ln -s /ifs1/User/wuqi/Qxy/knowngenome/orthofinder/Umbilicaria_antarctica.proteins.fasta ./ signalp -batch 30000 -org euk -fasta Umbilicaria_antarctica.proteins.fasta -gff3 -mature #运行速度还行,跑完后生成XX.gff3、XX_mature.fasta、XX_summary.signalp5文件结果文件XX.gff3是注释信息 XX.fasta是找到的分泌蛋白信号肽序列 XX.signalp5也是预测结果文件 11.2跨膜区分析,有跨膜区的会固定在膜上,无法成为分泌蛋白在线网站DeepTmhmm分析跨膜区,网址:https://dtu.biolib.com/DeepTMHMM(自己说的1和2不太行,现在推荐DeepTmhmm) 直接输入上一步得到的Umbilicaria_antarctica.proteins_mature.fasta然后run 运行结束后download下载gff3格式和3line格式,查看后感觉gff3格式中可以根据Number of predicted TMRs: 1来判断是否被固定在膜上,数为0则是分泌pr mkdir TMHMM cd TMHMM 将TMRs.gff3上传至服务器,运行下列代码 grep "Number of predicted TMRs: 0" TMRs.gff3 | perl -p -e 's/#\s+(\S+).*/$1/' > genes_without_TMHs.list ln -s ../singalp/Umbilicaria_antarctica.proteins_mature.fasta ./ grep -A 1 -f genes_without_TMHs.list -w Umbilicaria_antarctica.proteins_mature.fasta > Umbilicaria_antarctica.proteins_tmhmm.fasta #创建一个链接,然后从上一步singalp结果文件XX.mature.fasta中根据genes.list去得到tmhmm过滤后的分泌蛋白序列 11.3分析GPI锚定位点,GPI锚定蛋白和膜结合,从而固定到膜上,不会成为分泌蛋白 mkdir PredGPI cd PredGPI在线网站Predgpi直接进行预测,网址:https://busca.biocomp.unibo.it/predgpi/ (教材上给的网站都有误,自己扒拉了一个低配版) 但是!该网站一次只能运行100条序列,so需要将上一步得到的XXtmhmm.fasta文件传输到本地,手动分割一下序列,然后在Predgpi网站进行预测 速度较快,完成后可以在线查看预测结果,download下载csv格式(fasta格式下载为空!) 然后打开excel表,手动整理得到被预测为GPI-anchor的序列名称文件predGPI.list,传输到服务器,从Umbilicaria_antarctica.proteins_tmhmm.fasta中反向匹配删除#注!notepad++删除空行可以点击上方Edit编辑,选择line Operations,然后选择Remove Empty lines就可以啦! ln -s ../TMHMM/Umbilicaria_antarctica.proteins_tmhmm.fasta ./ grep -v -e "--" -f predGPI.list Umbilicaria_antarctica.proteins_tmhmm.fasta > Umbilicaria_antarctica.proteins_predGPI.fasta #创建一个链接,从上一步tmhmm结果中过滤得到no GPI的序列,并且去除--行 11.4进行亚细胞定位,选取定位到胞外的蛋白作为分泌蛋白基因 mkdir 4BUSCA cd 4BUSCA 将上一步XX.predGPI.fasta文件传输到本地,进入BUSCA在线网站进行分析,一次最多可以分析500条序列 网址:http://busca.biocomp.unibo.it/ ,选择序列文件,选择FUNGI类型,然后start prediction 运行完成后,将CSV文件传输到服务器 perl -e '; while () { @_ = split /,/; $stats{$_[2]}{$_[0]} = 1; } foreach (sort keys %stats) { @gene = sort keys %{$stats{$_}}; my $gene_number = 0; $gene_number = @gene; print STDERR "$_\t$gene_number\n"; if ($_ eq "C:extracellular space") { foreach (@gene) { print "$_\n"; } } }' BUSCA.out.csv > extracellular_gene.list 2> BUSCA.out.csv.stats ln -s ../3PredGPI/Umbilicaria_antarctica.proteins_predGPI.fasta ./ grep -A 1 -f extracellular_gene.list -w Umbilicaria_antarctica.proteins_predGPI.fasta | grep -v -- "--" > Umbilicaria_antarctica.proteins_BUSCA.fasta 最后得到的Umbilicaria_antarctica.proteins_BUSCA.fasta即为分泌蛋白的名称和序列,可以看看其相关功能等~最后拿到的.fasta文件就是想要的分泌蛋白基因啦!!! |


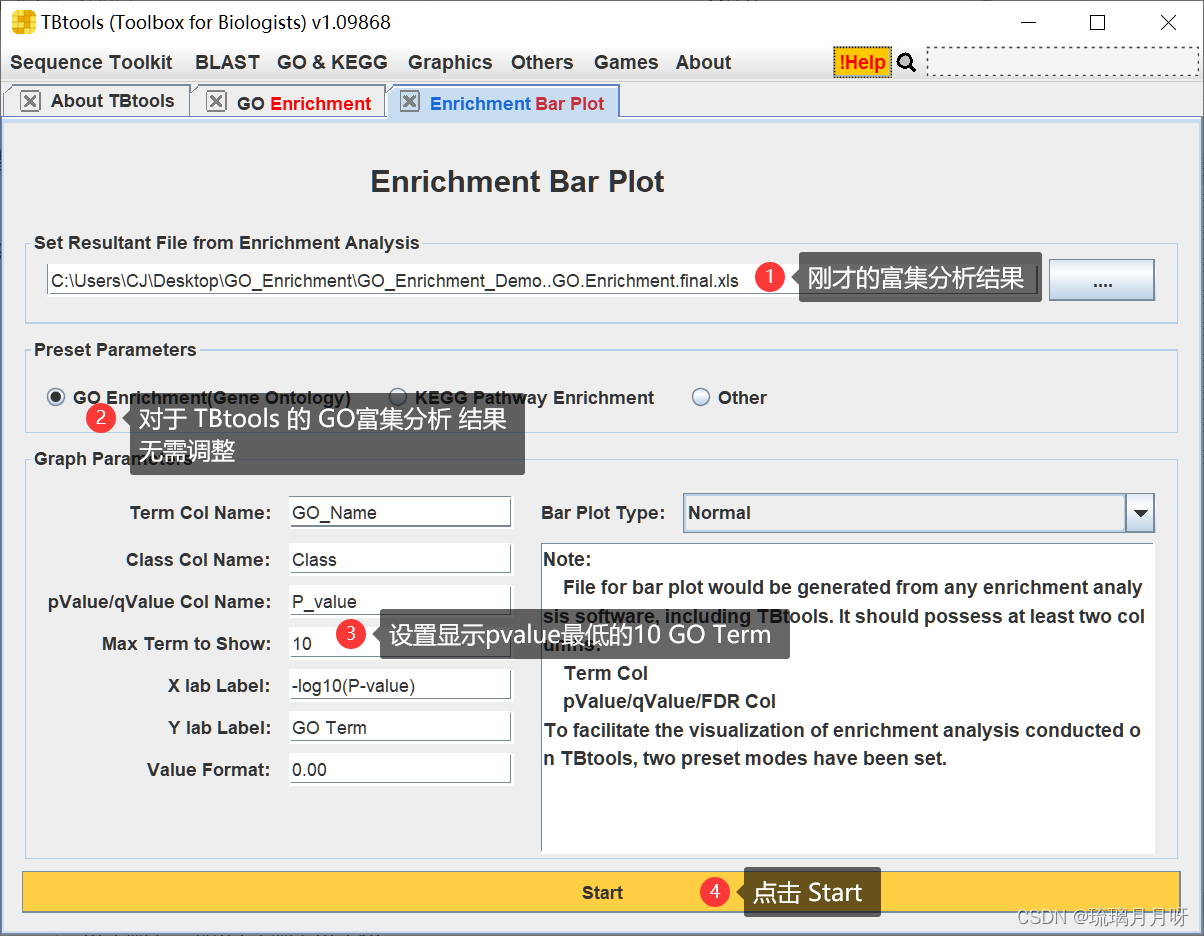 这里记得选pvalue最低的10个
这里记得选pvalue最低的10个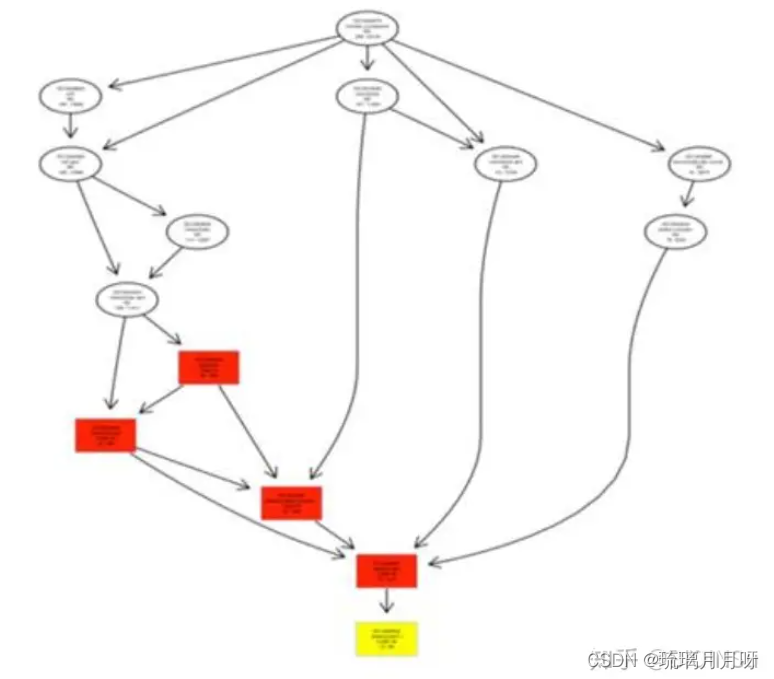
 这个图的意思是这么看的~
这个图的意思是这么看的~ 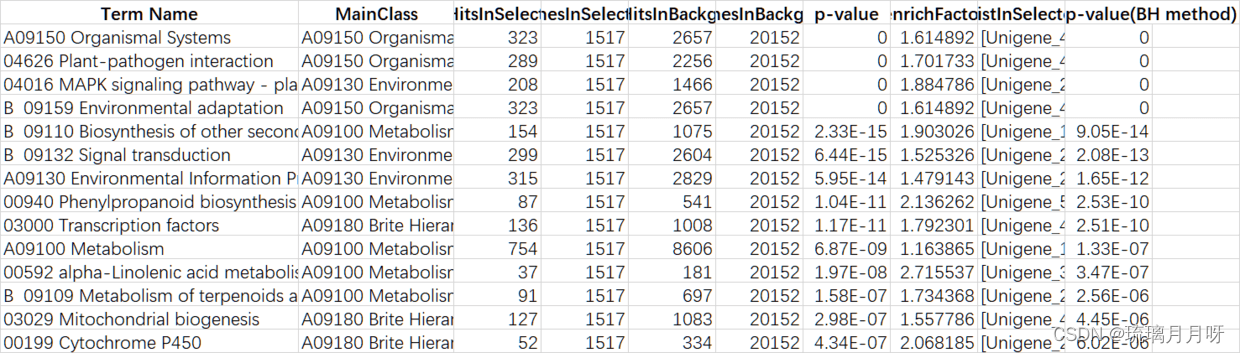
【本文地址】
今日新闻 |
推荐新闻 |