【学习记录】BLAST+简介、安装与使用 |
您所在的位置:网站首页 › ncbiblast怎么用 › 【学习记录】BLAST+简介、安装与使用 |
【学习记录】BLAST+简介、安装与使用
|
一、BLAST简介1.什么是BLAST BLAST是“局部相似性基本查询工具”(Basic Local Alignment Search Tool)的缩写,是由美国国立生物技术信息中心(NCBI)开发的一个基于序列相似性的数据库搜索程序,是目前最常用的数据库搜索程序。 BLAST实际是综合在一起的一组工具的统称,它不仅可用于直接对核酸序列数据库和蛋白质序列数据库进行搜索,而且可以将带搜索的核酸序列翻译成蛋白质序列后再进行搜索,或反之,以提高搜索效率。 常用BLAST分类: 标准BLAST:Blastn、Blastp、Blastx、tBlastn、tBlastx BLASTp根据搜索算法分:标准BLASTp、PSI-BLAST、PHI-BLAST 注:这里记录的是Blast+的简介、安装与使用,旧版的blast不完全适用。 2.BLAST是怎么工作的BLAST基本原理很简单,它的要点是片段对的概念。所谓片段对是指两个给定序列中的一对子序列,它们的长度相等且可形成无空位的完全匹配。 图 1-A 中方框里的就是两个片段对。BLAST 从头至尾将两条序列扫描一遍并找出所有片段对,并在允许的阈值范围内对片段对进行延伸,最终找出高分值片段对(high-scoring pairs, HSPs)(图 1-B)。这样的计算复杂度是 n 的一次方(n 是序列的长度)。如果做双序列比对话需要构建一个 n 乘以 n 的表格,计算复杂度是 n 的二次方。所以找高分值片段对比做双序列比对节省了大量的时间,当然,前提是牺牲了一定的准确度。另外,如今改进后的BLAST允许空位的插入。 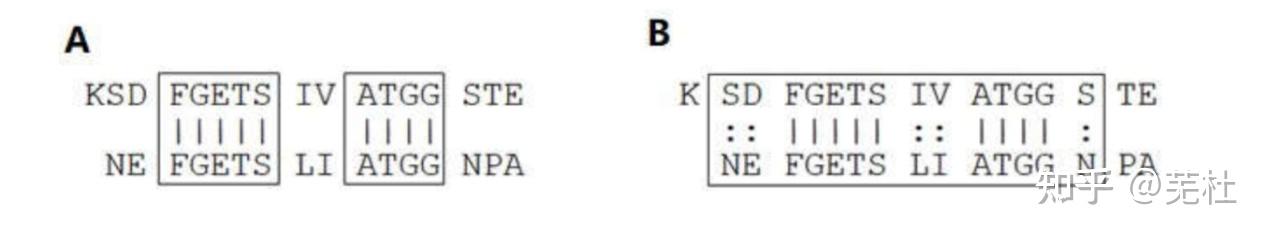 图1. 片段对及高分值片段对3.常用的BLAST程序介绍程序名查询序列数据库搜索方法Blastn核酸核酸将待查询的核酸序列及其互补序列一起对核酸序列数据库进行查询Blastp蛋白质蛋白质将待查询的蛋白质序列及其互补序列一起对蛋白质序列数据库进行查询Blastx核酸蛋白质先将待查询的核酸序列按六种可读框架(逐个向前三个碱基和逐个向后三个碱基读码)翻译成蛋白质序列,然后将翻译结果对蛋白质序列数据库进行查询tBlastn蛋白质核酸先将核酸序列数据库中的核酸序列按六种可读框架翻译成蛋白质序列,然后将待查询的蛋白质序列及其互补序列对其翻译结果进行查询tBlastx核酸核酸先将待查询的核酸序列和核酸序列数据库中的核酸序列按六种可读框架翻译成蛋白质序列,然后再将两种翻译结果从蛋白质水平进行查询 图1. 片段对及高分值片段对3.常用的BLAST程序介绍程序名查询序列数据库搜索方法Blastn核酸核酸将待查询的核酸序列及其互补序列一起对核酸序列数据库进行查询Blastp蛋白质蛋白质将待查询的蛋白质序列及其互补序列一起对蛋白质序列数据库进行查询Blastx核酸蛋白质先将待查询的核酸序列按六种可读框架(逐个向前三个碱基和逐个向后三个碱基读码)翻译成蛋白质序列,然后将翻译结果对蛋白质序列数据库进行查询tBlastn蛋白质核酸先将核酸序列数据库中的核酸序列按六种可读框架翻译成蛋白质序列,然后将待查询的蛋白质序列及其互补序列对其翻译结果进行查询tBlastx核酸核酸先将待查询的核酸序列和核酸序列数据库中的核酸序列按六种可读框架翻译成蛋白质序列,然后再将两种翻译结果从蛋白质水平进行查询为什么是按 6 条链翻译? 在无法得知翻译起始位点在情况下,翻译可能是从第一个碱基开始,三个三个的往后翻译,也可能是从第 2 个碱基开始,也可能从第 3 个碱基开始。另外还有可能是从这条链的互补链上开始,这样又有三个可能的开始位置,加起来一共会产生 6 条可能被翻译出来的蛋白质序列。这 6 条中有些是真实存在的,有些是不存在,但是谁真谁假我们无从知晓,所以 6 条序列都要到数据库中去搜索一下试试。 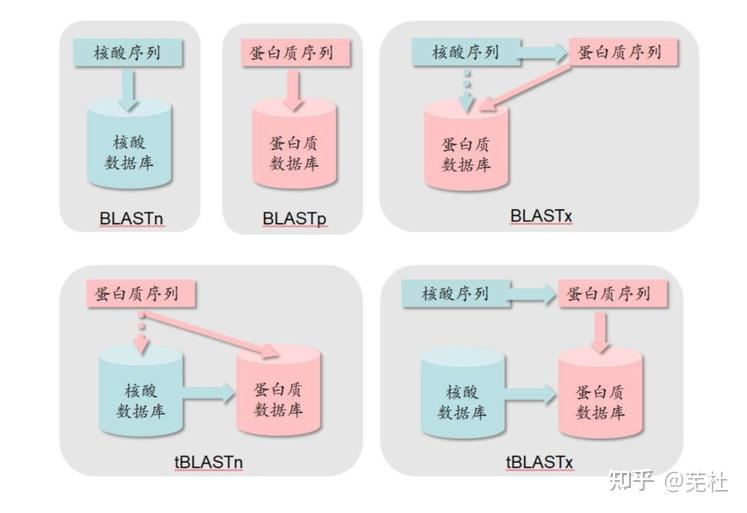 图2. 各种BLAST示意图4.PSI-BLAST和PHI-BLAST 图2. 各种BLAST示意图4.PSI-BLAST和PHI-BLAST①PSI-BLAST(Position-Specific Iterated BLAST,位点特异性迭代BLAST) PSI-BLAST的特色是每次用位置特异权重矩阵(Position-Specific Scoring Matrix,PSSM)搜索数据库后再利用搜索的结果重新构建PSSM,然后用新的PSSM再次搜索数据库,如此反复(iteration)直至没有新的结果产生为止。 ②PHI-BLAST(Pattern-Hit Initiated BLAST,模式识别BLAST) PHI-BLAST能找到与输入序列相似的并符合某种特定模式(Pattern)的序列,这种序列特征模式可能代表某个翻译后修饰的发生位点,也可以代表一个酶的活性位点,或者一个蛋白质家族的结构域、功能域。 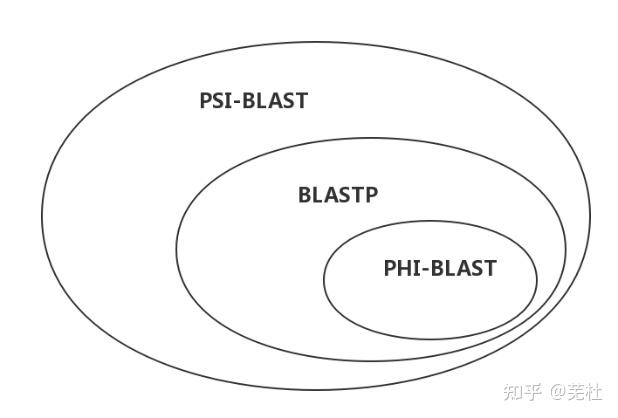 图3. PSI-BLAST、BLASTP和PHI-BLAST三者关系二、BLAST资源获取1.NCBI网址 图3. PSI-BLAST、BLASTP和PHI-BLAST三者关系二、BLAST资源获取1.NCBI网址网络版:https://blast.ncbi.nlm.nih.gov/Blast.cgi 单机版:ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST 2.版本比较(1)网络版 包括NCBI在内的很多网站都提供了在线的BLAST服务,这也是我们最经常用到的BLAST服务。网络版本的BLAST服务有方便,容易操作,数据库同步更新等优点。但是缺点是不利于操作大批量的数据,同时也不好自己定义搜索的数据库。另外分析时使用的服务器遥远,加上全球可能同时大量人员使用,所以使用NCBI的网络版BLAST,分析时间可能会很长,甚至会卡死中断。 (2)单机版 单机版的BLAST可以通过NCBI的ftp站点获得,有适合不同平台的版本(包括Linus,DOS等)。获得程序的同时必须获取相应的数据库才能在本地进行BLAST分析。单机版的优点是可以处理大批的数据,可以自己定义数据库,但是需要耗费本地机的大量资源,此外操作也没有网络版直观、方便,需要一定的计算机操作水平。 3.本地WEB版的BLAST使用如viroBLAST和Sequenceserver(http://www.sequenceserver.com)软件包,用户可以建立一个简易的进行BLAST运算的网站供实验室人员使用,该网站相当于NCBI-BLAST(ncbi-blast+)的前端图形交互界面。 4.BLAST程序评价序列相似性的两个数据(1)Score:使用打分矩阵对匹配的片段进行打分,这是对各氨基酸残基(或碱基)打分求和的结果,一般来说,匹配片段越长、相似性越高则Score值越大。 (2)E-value:在相同长度的情况下,两个氨基酸残基(或碱基)随机排列的序列进行打分,得到上述Score值的概率的大下。E值越小表示随机情况下得到该Score值的可能性越低。 三、BLAST的安装与使用1.本地BLAST的安装(1)Windows平台 ①程序下载: 访问BLAST本地软件包下载链接(ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST),下载适合自己电脑的BLAST win版本。 ②安装: 下载完毕后,双击安装到指定位置(如:C:\Blast),生成bin和doc两个子目录,其中bin是程序目录, doc是文档目录,这样就安装完成。 ③环境变量设置: 右键点击“我的电脑”-属性,然后“高级系统设置”选项-“环境变量”,在用户变量下方点击“新建”-变量名:BLASTDB,变量值:C:\Blast\db(即数据库路径)。在系统变量下方“Path”添加变量值:C:\Blast\bin。 ④查看程序版本信息: 点击window的"开始"菜单,在运行中输入cmd,调出MS-DOS命令行,输入命令cd C:\Blast\bin转到BLAST安装目录,输入命令 blastn -version即可查看版本。 注:以上涉及的地址需要按照自己电脑BLAST安装的位置进行设定。 (2)Linux平台 ①下载: wget ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ncbi-blast-2.13.0+-x64-linux.tar.gz②解压: tar -zxvf ncbi-blast-2.13.0+-x64-linux.tar.gz③重命名(为方便使用和统一管理): mv ncbi-blast-2.13.0+-x64-linux blast④环境变量设置: 将BLAST+可执行程序所在目录(bin)的绝对路径加入到环境变量$PATH中,以方便通过程序名直接调用。 编辑~/.bashrc文件,在最后加入以下行: export PATH=/home/local/Software/blast/bin:$PATH如果不会使用vi/vim等编辑器,可直接运行下列一行命令,将上述内容添加到~/.bashrc文件(看清楚,和上面不同的是:$被转义了的): echo "export PATH=/home/local/Software/blast/bin:\$PATH" >> ~/.bashrc⑤让配置生效: source ~/.bashrc 到此,你就可以直接输入BLAST的子程序,如blastn进行比对了。输入blastn –version可查看版本信息。 以上涉及地址需要按照自己电脑BLAST安装位置进行设定,由于NCBI会更新BLAST+,所以以上命令在下载时需要查看当时的版本信息而进行相应的修改。 2.本地BLAST的使用(1)数据格式化(检索库的建立) ①格式化命令: makeblastdb -in db.fasta -parse_seqids -hash_index -dbtype prot②主要参数说明: -in参数后面接将要格式化的数据库; -parse_seqids, -hash_index两个参数一般都带上,主要是为blastdbcmd取子序列时使用; -dbtype 后接所格式化的序列的类型,核酸用 nucl,蛋白质用prot; -title 给数据库起个名(不能用在后面搜索时-db的参数); -out 后接数据库名,自己起一个有意义的名字,后面比对搜索时要用到的-db的参数; -logfile 日志文件,如果没有默认输出到屏幕。 (2)BLAST序列比对 ①序列比对命令: 例:核酸序列比对蛋白数据库: blastx -query test.fasta -out test.blast -db db.fasta -outfmt 6 -evalue 1e-5 -num_threads 2蛋白序列比对蛋白数据库: blastp -query test.fasta -out test.blast -db db.fasta -outfmt 6 -evalue 1e-5 -num_threads 2②主要参数介绍: -query: 输入文件路径及文件名; -out:输出文件路径及文件名; -db:数据库路径及数据库名; -task: 共五个程序选择'blastn' 'blastn-short' 'dcmegablast' 'megablast' 'rmblastn' ,默认megablast; 具体区别如下: blastn 完全匹配的传统blastn; blastn-short 优化查询:短于50个碱基; megablast 查找十分相似的序列(如物种内部或相关的物种间); dc-megablast 查找亲缘关系比较远的序列(如物种间); rmblastn 兼容了RepeatMasker。 -evalue:设置输出结果的e-value值,一般1e-5; -num_threads:线程数,笔记本不要设大了,2就够了; -num_alignments:输出数据库中能与Query比对上的的序列数目,与max_target_seqs不能同时使用; -max_target_seqs:最多允许比对到数据库中的序列数目,参数仅适用于outfmt >4; -perc_identity :比对的最低相似度; -max_hsps:由于不对时一条序列比对成多段,如果只想输出其中的几段,就设定相应的数目,与-num_alignments不能同时使用; -outfmt:输出文件格式,总共有15种格式,一般设置为6。6是tabular格式对应BLAST的m8格式; 0 = pairwise, 1 = query-anchored showing identities, 2 = query-anchored no identities, 3 = flat query-anchored, show identities, 4 = flat query-anchored, no identities, 5 = XML Blast output, 6 = tabular, 7 = tabular with comment lines, 8 = Text ASN.1, 9 = Binary ASN.1, 10 = Comma-separated values, 11 = BLAST archive format (ASN.1), 12 = JSON Seqalign output, 13 = JSON Blast output, 14 = XML2 Blast output。 此外还能自定义输出格式主要针对上述的 6, 7, and 10三种格式。 ③outfmt 6输出12列对应的含义: Query id:查询序列ID标识; Subject id:比对上的目标序列ID标识; % identity:序列比对的一致性百分比; alignment length:符合比对的比对区域的长度; mismatches:比对区域的错配数; gap openings:比对区域的gap数目; q. start:比对区域在查询序列(Query id)上的起始位点; q. end:比对区域在查询序列(Query id)上的终止位点; s. start:比对区域在目标序列(Subject id)上的起始位点; s. end:比对区域在目标序列(Subject id)上的终止位点; e-value:比对结果的期望值,解释是大概多少次随即比对才能出现一次这个score,Evalue越小,表明这种情况,从概率上越不可能发生,但是现在发生了,所以这个比对具有很重要的意义; bit score:比对结果的bit score值。bit score值表示对齐的好坏;分数越高,对齐效果越好。一般来说,这个分数是根据一个公式计算出来的,这个公式考虑了相似或相同残基的排列,以及为了排列序列而引入的任何gaps。 3.网络版BLAST的使用(1)序列比对 ①进入BLAST网页,选择需要使用的BLAST   ②输入查询序列 除了可以输入单条序列,还可以通过上传文件进行多条序列的比对。 ③选择搜索的数据库  (2)比对结果查看 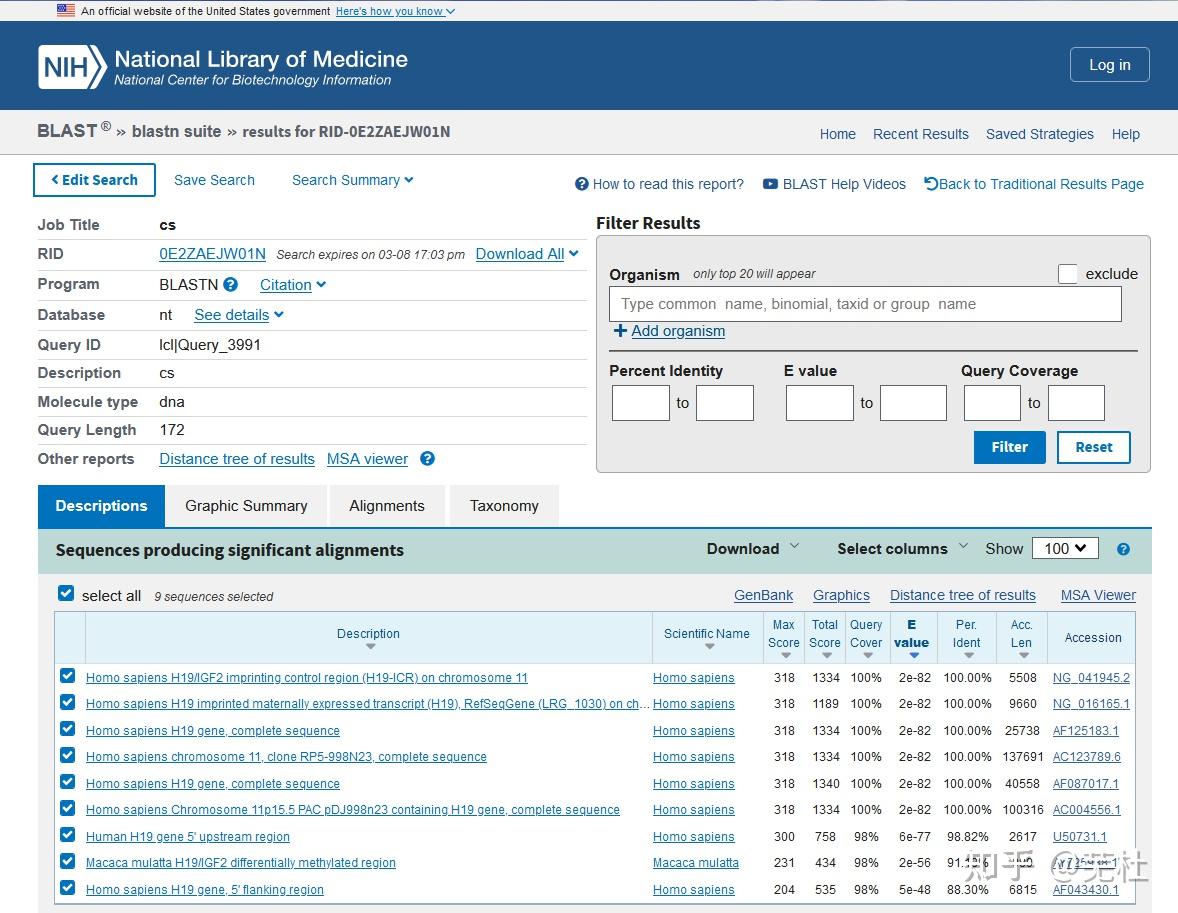 第一部分搜索任务描述部分。输入界面里设置的各种参数都会在这里列出。 第二部分(Descriptions)是高分匹配片段所在序列的详细信息列表。每条序列都有一个匹配得分和覆盖度。这两项决定了第二部分彩图中每条线的颜色和长短。除了匹配得分和覆盖度,表中还列出了其他指标。尤为重要的是 E-value。E-value 也叫做期望值或 E值。E 值越接近零,说明输入序列与当前这条序列为同一条序列的可能性越大。第三部分的表就是根据 E 值由低到高排序的。随着 E 值增大,匹配得分是成反比逐渐降低的。但是一致度与 E 值并非完全成反比。因为我们在前面讲 BLAST 核心思想的时候说过,BLAST 没有做双序列比对,为了提高速度,它牺牲了一定的准确度。表中的一致度,是 BLAST 搜索完成后,针对搜索到的这几条序列专门做双序列比对而得到的。BLAST 牺牲掉的准确度对高度相似的序列,也就是亲缘关系近的序列构成不了威胁,不会把它们落掉,但是对于那些只有一点点相似,也就是远源的序列,就有点麻烦了,它们很有可能被丢掉而没有被 BLAST发现。 第三部分(Graphic Summary)是图形化搜索结果部分。在图形化搜索结果里,BLAST结果图形显示相似度颜色图(黑、蓝、绿、粉红、红,相似度由低到高)。 第四部分(Alignments)是比对的详细信息。 第五部分(Taxonomy)是比对上的物种分类学信息。 |
【本文地址】
今日新闻 |
推荐新闻 |