经验分享:线粒体mRNA基因变异的解读 |
您所在的位置:网站首页 › mtdna中编码基因的数目为 › 经验分享:线粒体mRNA基因变异的解读 |
经验分享:线粒体mRNA基因变异的解读
|
人类的mtDNA基因组包括16569个碱基对,一共编码37个基因,包括2个核糖体RNA,22个线粒体转运RNA(mt-tRNA)和13个蛋白质编码线粒体信使RNA(mt-mRNA)[5]。由于tRNA的结构与线粒体生物学特点,对于mt-tRNA变异的解读十分独特[6]。mt-mRNA基因编码7个复合体I亚基(MT-ND1, MT-ND2, MT-ND3, MT-ND4, MT-ND4L, MT-ND5和MT-ND6),1个复合体III亚基(MT-CYB),3个复合体IV亚基(MT-CO1, MT-CO2和MT-CO3)和2个复合体V亚基(MT-ATP6和MT-ATP8)[7]。对mt-mRNA变异的分类解读基于核基因组mRNA基因的解读标准,同时需结合mtDNA特征进行改进和优化。 在本研究中,我们回顾了已报导的mt-mRNA变异以提取分类规则。随后,我们用内部数据库中已报导变异的新病例对这些规则进行完善,并将完善后的规则应用于VUS和新变异的分类。 方法mt-mRNA变异的来源 本研究基于Baylor College of Medicine 和Baylor Genetics 的线粒体分子诊断实验室的经验。在>10,000个mt-DNA变异中,除去高频mtDNA SNP(人群频率>5%)以及非编码区、rRNA和tRNA区域中的变异,筛选出约4800个罕见或新的mt-mRNA变异,其中3005个是同义变异,1795个是非同义变异(图1,见文末)。 mt-mRNA变异解读标准的测试和完善 我们将1795个罕见的mt-mRNA变异分为两组:421个新变异和1374个文献或公共数据库报导过的变异。基于已报导的情况,后者又分为三类:致病(P)变异,致病性未明变异(VUS)和良性(B)变异(图1)。我们系统回顾了已报导的1374个mt-mRNA变异的致病性证据,根据ACMG指南[3]同时结合线粒体特征,进而从具有足够证据的P和B变异中推导出分类规则(图1;附表S1,“添加新病例前的评估”列)。然后,我们使用新病例检验和完善了这些标准(附表S1,“添加新病例后重新评估”列)。随后,我们用经过微调和验证的标准去重新评估已报导的没有充分证据的变异。然后再应用已建立的标准(表1和2)去重新评定已报导的VUS和新变异(图1)。变异的分类联合标准同ACMG指南[3]。
B良性的,LB 可能良性的,LP可能致病的,mtDNA 线粒体DNA,mRNA 信使RNA,rRNA 核糖体RNA,P 致病的,SNP单核苷酸多态性,VUS 致病性未明变异。
左列为ACMG分类标准,中间为mt-mRNA变异的新分类标准,右列为mt-tRNA变异的新分类标准。ACMG 美国医学遗传学与基因组学学院,ATP 三磷酸腺苷,COX 细胞色素C氧化酶,ETC 电子传递链,mt-mRNA 线粒体信使RNA,mt-tRNA 线粒体转运RNA, NGS 二代测序,OCR 耗氧率,PCR 聚合酶链式反应。 结果评估已报导的mt-mRNA变异 我们从临床诊断实验室筛选出了1795个罕见的mt-mRNA变异,并将这些变异提交到了ClinVar数据库(https://www.ncbi.nlm.nih.gov/clinvar/),检索号为# SCV000996654至SCV000998448。在1795个变异中,有421个变异是新变异,其他1374个变异有56个评定为P,1111个评定VUS,207个评定为B(图1,见文末)。经文献查找发现56个P变异中有32个有充分的证据证明其致病性。致病依据主要有:与先前的明确致病变异有相同的氨基酸改变(PS1),线粒体功能损伤程度与变异的异质率相关(PS3),无效变异(PS6),变异异质性≥10%且变异异质率与患者不同组织受累的临床表型相关(PM8),变异异质率与不同母系家庭成员或不同家系的患者临床表型相关(PM9)。与既往文献中分类不一致的变异可能是由于缺乏一个或者多个上述致病依据(13,附表S1,见文末)。 P变异降级为LP P变异降级为LP最常见的原因是缺乏功能研究证据(PS3),而且之前的变异分类为三级分类(分为 P、VUS和B),其中没有LP类别。例如,m.8851T>C (p.W109R, MT-ATP6)变异之前报导于一名2.5岁患有双侧纹状体坏死的男童和一名3岁患有Leigh综合征的女童 (PP4) [8.9]。该变异在先证者中表现为较高的异质率(>85%),而在未受影响的母亲中表现为较低的异质率(68%)(PP6, PM9) [9]。尽管肌肉病理学和ETC结果支持致病性,但功能受损程度与变异异质率的相关性不明(PM10)。SIFT和PolyPhen2软件预测该变异对基因或蛋白功能有害(PP3)。因此,该变异被重新评定为LP变异 (2PMs, 3PPs)。我们在一例53岁患有脑病、肌病和中风的女性患者血液中检测出接近同质水平的m.8851T>C;在一例临床表型为大脑皮质发育异常的2岁男孩中检测到了同质性的m.8851T>C变异。此外,我们在一例36岁的女性中检测到同质性m.8851T>C变异,且该女性同时还具有异质率为75%的m.8993T>G变异。由于缺乏该变异异质率与功能损伤程度相关的研究,且不同患者的临床表型存在较大差异,我们将该变异评定为LP。 M.13046T>C (p.M237T, MT-ND5)变异曾在一例患有Leber遗传性视神经病(LHON)/线粒体脑肌病伴乳酸中毒及中风样发作(MELAS) (PP4)的患者中报导过。该变异在血液、肌肉和尿液中的异质率分别为22%、50%和52%(PM8, PP6)[10]。在另一例LHON/MELAS患者的肌肉和血液中检测到该变异异质率分别为70%和27%,而在无症状母系亲属的多种组织中未检测到该变异(PM9) [11]。线粒体复合体I的活性在肌肉组织中中度减少(50%),不满足线粒体病的诊断标准,且两篇文献均未做功能研究,因此该变异仅被认定为可能致病变异(LP)(表3)。尽管我们还在一个无症状婴儿的血液中发现了该变异,其异质率仅为3.2%,此新病例这既不能支持也不能反驳m.13046T>C的致病性,故仍将其评定为LP(2PMs, 2PPs)。 M.13063G>A (p.V243I, MT-ND5)变异曾在一名临床表型为共济失调和睡眠性肌阵挛(PP4) 的32岁患者中报导过,其异质率在肌肉和外周血中分别为80%和25%(PP6, PM8)。虽然线粒体复合体的活性在转线粒体胞质杂交细胞(transmitochondrial cybrids)中与变异异质率相关(PS3),但该变异在部分无症状的母系亲属的淋巴细胞中被检测出超过25%的异质率[12]。此外,我们在携带PYGM基因分子缺陷的肌病患者的肌肉中也检测到该变异,其异质性为17.9%。我们的案例既不能支持也不能反驳它的致病性。因此,由于证据不足,这个变异仍然评定为LP。 P变异降级为VUS 因为缺乏功能实验或变异异质率与表型的相关性,7个已报导的P变异被降级为VUS(表3)(缺乏PS3和/或PM8/PM9)。其余的变异可能在数据库中被报导为多态性(BS1), 或者拥有矛盾的证据如在无症状母亲中表现为同质性(BS4)。例如,m.9029A>G (p.H168R, MT-ATP6)之前在LHON患者中被报导为同质性变异(PP4) [13],其在患者的无症状的母亲(95%)和兄弟姐妹(85%和95%)中也接近同质性(PM9)。转线粒体胞质杂交细胞研究未能证实线粒体功能的降低与变异异质率相关(PM10)。我们在两名脑病患者及其健康母亲的血液中检测到了同质水平的m.9029A>G。这些不一致的证据对m.9029A>G的致病性提出了质疑(2PM, 2PP, 1BS)。 在73名疑似线粒体疾病的儿童患者(无详细临床资料)中,发现2名患者携带异质性的无义变异m.9984G>A (p.G260*, MT-CO3)[14]。MT-CO3的第260位密码子为倒数第二个密码子,只有当这两个末端氨基酸对蛋白质的结构/功能至关重要时,此处发生的变异才会导致临床表现。此外,我们在临床症状为顽固性癫痫的15岁女性患者血液中检测到了该变异,其异质率为7.8%。尽管无效变异会导致蛋白截短的产生或者全部编码蛋白缺失,但低异质率的无效mt-mRNA变异可能不具有致病性。因此,我们的新病例既不支持也不能反驳该变异的致病性。由于证据不足,我们将m.9984G>A降级为VUS(1PP)。
B良性的,LB 可能良性的,LP 可能致病的,P致病性的,VUS 致病性未明变异 a m.4142G>A 是唯一升级到P的VUS P变异降级为LB和B 在公共数据库中,发现了6例先前报导的缺乏功能实验和/或异质性研究的P变异(BS2)。并且在我们内部数据库中,这些变异在无症状母系亲属中被检出(BS4),和/或被多种软件预测为良性(BP4)。在重新评估后,我们将3个P变异降级为LB,另3个P变异降级为B。例如,m.9957T>C (p.F251L, MT-CO3)变异首次在MELAS患者中被报导。该变异在先证者及其无症状母亲的血液中具有相似的异质率。肌肉病理中未观察到细胞色素c氧化酶(COX)缺陷,肌肉匀浆中复合体的活性没有受损[15]。随后,该变异在多个具有不同临床表型的家系中被报导,包括肥厚型心肌病[16]和非动脉炎性缺血性视神经病[17]患者,以及痛风的成年男性[18] 。在受累和无症状的家庭成员中m.9957T>C可检出,异质率与临床表型无相关性。转线粒体胞质杂交细胞研究显示线粒体ROS生成增加,但线粒体呼吸链复合体酶的活性或膜电位未发生重大变化[19]。该变异在MITOMAP(https://www.mitomap.org/)中出现了四十次,在线粒体数据库(mtDB, ) http://www.mtdb.igp.uu.se/ 中出现了一次,在我们的数据库的健康母亲群体中出现了两次以上。此外,预测软件提示该变异为良性(BP4)。因此,我们将m.9957T>C变异降级为良性变异(B)。从P变异降级为B的变异还包括m.13528A>G (p.T398A, MT-ND5;BS1, BS2, BP4) 和m.15804T>C (p.V353A, MT-CYB;BS1, BS4, BP4, BP6)。从P变异降级为LB的变异包括m.3340C>T (p.P12S, MT-ND1)、m.3395A>G (p.Y30C, MT-ND1)和m.12622G>A (p.V96I, MT-ND5)。降级原因主要包括:已被报导为多态性(BS1),软件预测为良性变异(BP4),以及在多个健康的母系家庭成员或无血缘家庭中表现为同质性水平(BS4/BP6)。 VUS升级为LP 我们使用已完善的规则重新评估了1111例已报导的VUS(附表S1)。根据变异在不同数据库中出现的频率和在无症状母亲或健康成人中的情况,大多数(65.7%)被评定为LB(299/1111=26.9%)或B(431/1111=38.8%),34.2%(380/1111)仍然为VUS。只有1例VUS升级为LP(表3)。变异m.4142G>A (p.R279Q, MT-ND1)出现在既往报导中但未被分类[20]。我们在一名临床表型为发育迟缓、肌张力减退、中风、癫痫、大头畸形和乳酸升高的10个月龄男孩肌肉中检测到该变异,其异质率约为80%。而在其无症状母系亲属中未检测到该变异(PM9)。我们还在一名临床表型为癫痫和中风的12岁女孩的肌肉和血液中检测到该变异,其异质性分别为65.3%和31%(PM8, PP4, PP6)。该变异同样未在其无症状的母亲和母系亲属身上检测到,并且在MITOMAP或mtDB中也未收录该变异(PM2)。因此,我们将m.4142G>A 变异评定为LP (3PMs, 2PPs)。 B变异升级为LB 我们重新评估了先前报导的207例B变异。几乎所有变异仍然是B(204/207),只有3例被重新评定为LB(3/207,这是因为它们在公共数据库中出现的频率相对较低,或者在我们的数据库中没有多个健康母系亲属或独立家系的数据支持。这三个变异为m.7362G>A (p.E487K, MT-CO1)、m.7363A>G (p.E487G, MT-CO1)和 m.14564A>G (p.V37A, MT-ND6)(见附表S1)。 新变异的解读 我们发现了421个新变异,其中2个被评定为P变异,12个被评定为LP(表4),384个被评定为VUS,23个被评定为LB,0个被评定为B(图1,见文末,附表S1)。其中有8个新的无效变异(无义变异或移码变异)被评定为P或LP变异。例如,在一名临床表型为发育迟缓、癫痫和心肌病的患者肌肉样本中检测到m.5001dupA(p.I178Nfs*22,MT-ND2)这一无效变异,其水平接近同质(PS6,PP4)。而在他母亲中未检测到该变异,且该变异在公共数据库中没有被报导过(PM9,PM2)。功能研究表明线粒体呼吸链复合体I和复合体I+III的活性均降低(分别为对照组的46%和47%)。线粒体功能受损还体现在线粒体增殖,表现为柠檬酸合酶活性增加及mtDNA含量增加(为对照组的153%)(PM10)。因此,m.5001dupA被评定为P变异(1PS,3PM,1PP)。
在一名患有发育迟缓和肌病(PP4)的患者中发现了m.8993_8994TG>CA (p.L156P, MT-ATP6)变异,通过对先症者血液Sanger测序,发现该变异接近同质性(PP6),但在其无症状的母亲中并没有检测此变异,且该变异在公共数据库中没有被报导过(PM2,PM9)。更重要的是,该变异引起了与m.8993T>C (PS1)相同的氨基酸改变p.L156P。因此这个变异被评定为P(1PS, 2PMs, 2PPs)。 根据变异的异质率与表型的相关性(PM8/PM9)、和/或功能研究(PM10)、生信预测(PP3)、以及多个家系中相似的临床表型(PP4),5个新的错义变异被评定为LP。例如,我们在4个家系中检测到了m.14465G>A (p.T70I, MT-ND6)。第一个病例是一个9岁女孩,其临床症状表现为发育迟缓,肌张力减退,脑病,乳酸酸中毒,以及小头畸形。m.14465G>A在肌肉中为同质性水平。第二个病例是一个患有代谢紊乱、四肢瘫痪、大动脉移位、全身迟缓、退化、便秘和胃排空迟缓的2岁男孩,其血液中检测到了同质性的m.14465G>A变异。与其不同的是,该变异在他的妹妹中异质率为73.9%,而在他的无症状母亲血液中约为50% (PM2,PM9,PP4,PP6)。第三个病例是一个11岁女孩,患有发育迟缓、共济失调和乳酸酸中毒。m.14465G>A变异在先证者血液中为92.8%,而在她的无症状母亲身上没有检测到该变异。根据收集到的证据(2PMs,2PPs),我们将m.14465G>A评定为LP变异。 我们在一例临床表型为发育迟缓、肌张力低、肌病和发育不全的18岁女性中检测到新变异m.6526T>C(p.M208T,MT-CO1) (PP4,PP7),该变异异质率在先证者的肌肉中为86.6% (PP6)。肌肉mtDNA含量高达256%,表明线粒体增殖,结合ETC指标在增殖校正前后均满足线粒体呼吸链复合体IV缺乏症的主要标准(对照组的20%)(PM10)。多种软件预测该变异是有害的(PP3)。因此,m.6526T>C被评定为LP变异(1PM, 4PPs)。 当新变异是同质性的,在无症状的成年母系亲属中出现两次以上(BS4),且多种软件预测该变异是良性的(BP4),则这些变异可以被评定为LB。在新变异中, LB变异的数量很少(23/421=5.4%),且没有被评定为B的变异。这是由于绝大多数B变异早已被发现,且在大规模的测序数据库中因为频率过高而被过滤掉了。 讨论评估mt-mRNA基因变异致病性的独特因素 我们将核基因变异解读的规则和线粒体基因组的特征相结合,用以解读mt-mRNA基因变异。表1和表2将核基因和线粒体基因变异解读的标准进行对比。尽管在核基因的变异解读指南中,无效变异是一条非常强的致病性证据(PVS1), 但在mt-mRNA变异致病性的评定中,考虑到异质率及阈值效应的不确定性,以及核基因对表型差异的影响,对无效变异有必要降低其致病性评级证据等级。此外,无义变异可能因为位于蛋白C-末端附近而致病性弱,例如上文所述的m.9984变异。因此,我们为异质率≥10%的无效变异创建了PS6证据。mt-mRNA变异异质率的切割值定为10%,而不是mt-tRNA的5%,这主要是由于mt-mRNA变异仅导致某个蛋白质中的某个氨基酸的改变,而mt-tRNA变异会导致所有线粒体编码的蛋白质中由这个tRNA转运的氨基酸的异常。值得注意的是,异质率通常不是LHON相关mt-mRNA变异需考虑的因素,因为原发变异(primary mutations)通常是同质性的或接近同质性的。影响LHON变异的因素包括外显率,核基因组和/或单倍群背景对表型的影响,以及mtDNA协同二级变异和环境的影响。此外,mt-mRNA的LHON变异在受累人群中频率明显高于未受累人群[21.22],这种情况可考虑使用PS4证据。 mt-tRNA和mt-mRNA致病变异分布的比较 mt-tRNA基因仅占整个线粒体基因组的9.08%(150/16,569),而mt-mRNA基因约占68.45%(11341/16569)(表5)。事实上,mt-mRNA的总编码区长度大约是mt-tRNA序列的7.54(11341/1504)倍。然而,mt-tRNA基因每100bp中被评定为P和LP的变异总数(69/15.04=4.59)是mt-mRNA基因(58/113.41=0.51)的~9倍,表明mt-tRNA变异比mt-mRNA变异更具危害性。在既往已报导的变异中,mt-mRNA基因P变异P被降级到B/LB变异的概率(6/56)高于mt-tRNA基因 (4/72),进一步证实了上述观点。这可能是由于tRNA在加工过程中酶的识别和结构配对方面受到了严格的限制,并且mt-tRNA序列的变异会影响所有(13个)mtDNA编码蛋白的翻译。mt-tRNA新变异中P和LP变异的密度是mt-mRNA的5.5倍(tRNA/mRNA = 0.66/0.12 = 5.5),该比值远低于其在已报导变异(10.05=3.92/0.39)或总变异(9.00=4.59/0.51)中比值。与此不同的是,当考虑同义变异时,VUS、B和LB变异的tRNA/mRNA密度比值接近于1,这主要是由于贯穿了整个线粒体基因组的相等的变异几率所引起的。
mt-mRNA 线粒体信使RNA,LP 可能致病的,P 致病的 我们用改进后的新标准重新审核了本研究中所有的P和LP变异,同时也重新评估了MITOMAP数据库中所有报导为P的变异。我们重新评定出120个分类为P/LP的mt-mRNA变异(附表S2),以及160个分类为P/LP的mt-tRNA变异。一般来说,mt-tRNA 的P/LP变异比mt-mRNA的P/LP变异能引起更严重的表型,并且阈值较低。 LHON变异的解读 LHON的临床特征是视网膜神经节细胞层和视神经变性,导致双眼中央视力受损[23]。LHON通常发生在青春期或成年早期,男性发病率明显高于女性[24]。大约90%的病例是由线粒体呼吸链复合体I亚基中的三个变异m.3460G>A(p.A52T, MT-ND1)、m.11778G>A(p.R340H, MT-ND4)和m.14484T>C(p.M64V,MT-ND6)中的一个引起的[25]。与其他异质性致病变异不同,LHON变异的遗传特性通常具有不完全外显性,并且LHON变异在受累和未受累的母系亲属中都是同质性的[26]。这些独特的特征使得对新LHON变异的解读变得困难,因为与异质率相关的PM8和PM9证据对于LHON变异不适用。此外,由于不完全外显性,在无症状个体中常见同质性变异。在三个最常见的LHON变异中,只有m.3460G>A变异导致线粒体呼吸链复合体I的活性显著降低,另外两个变异对复合体I活性的影响相对较小。因此,常规线粒体呼吸链酶复合体的结构与功能研究可能无法验证LHON变异的致病性。 MITOMAP数据库收录LHON 相关P变异共18个,其中11个用我们的标准可证实其致病性。三个最常见的LHON变异的致病性可通过转线粒体胞质杂交细胞研究证明,且LHON患者中这三个变异的携带率高于正常对照人群。导致LHON伴其他线粒体病症状的异质性变异解读起来更容易些。例如,m.13094T>C(p.V253A,MT-CO1)变异在1名共济失调和进行性外眼肌麻痹(PEO)的患者中首次被报导[27]。该变异在患者的肌肉、淋巴细胞和成纤维细胞中检测出异质率分别为50%、40%和30%,但在其母亲中并未检测到。转线粒体胞质杂交细胞研究证实了异质率与线粒体呼吸链复合体I活性的相关性。后来,在一名LHON患者的血液中也发现了m.13094T>C变异, 其异质率为26%[28]。 然而,LHON的同质性罕见变异的致病性很难证明。例如,m.4171C>A(p.L289M,MT-ND1)变异在两个韩国LHON家系[29]中被首次报导。在第一个家系中,该变异在受累患者中为同质性,但在其健康母亲中检测到异质率为88%。在另一个家系中,该变异在表型为LHON和Leigh综合征的16岁男性先证者和他的一个无症状姐妹中被检测到,且均为同质性[30]。此外,我们还在一名视神经萎缩患者中发现了该变异。由于缺乏功能研究,且在无症状的母系亲属中存在明显的同质性,以及不同家系的表型不一致,我们将该变异降级为LP变异。 LHON变异的不完全外显性和性别偏倚提示有其他的遗传和/或环境因素参与了发病过程。有人提出单倍群 [31.32]和核基因组遗传背景[21]也会影响外显率。进一步了解LHON的其他遗传因素,以及建立更适合LHON变异的功能研究,可能更有助于LHON变异的分类解读。 mt-mRNA变异的基因型与表型相关性 正如附表S2中所总结,在120个mt-mRNA的致病变异中,线粒体复合体I占54个,线粒体复合体III占19个,线粒体复合体IV占26个,线粒体复合体V占21个。P和LP变异在线粒体复合体V的基因中密度最高(21/8.42 = 2.49,表6),其次分别是复合体III(1.67),复合体IV(0.86)和复合体I(0.85)的基因。线粒体复合体I致病变异密度低可能是由于复合体I有很多的亚基,因此单个亚基中的变异对整个复合体I影响相对小。线粒体复合体IV功能障碍在核基因编码的线粒体复合体组装基因(如SURF1基因)的变异最常见。线粒体复合体V的变异几乎都在MT-ATP6中。仅一个致病的无义变异(m.8529G> A,MT-ATP8中的p.W55 *和MT.ATP6中的p.M1M)和一个错义变异(m.8528T> C,MT-ATP8中的p.W55R,和 MT-ATP6中的p.M1T)位于MT-ATP8/MT-ATP6的重叠区域。P/LP的错义变异没有在MT-ATP8中被单独发现过。我们先前报导过变异m.8528T> C会引起婴儿型心肌病[33]。此后,我们还发现了另外四个家系,表明这个变异可能是引起婴儿型心肌病比较常见的原因。 在120个P/LP变异中,有75个是错义变异,40个是无义变异或移码变异,一个是七个核苷酸的倒置[34],另一个是在MT-ATP6的终止密码子处有2 bp的缺失(m.9205delTA,p.* 227MextX282[35]),三个是框内缺失(in-frame deletions)。有趣的是,无效变异的分布在不同基因之间完全不同。线粒体复合体I中,只有17%(9/54)的P/LP变异为无效变异,其中四个在MT-ND2基因里。19个MT-ATP6基因变异中的六个变异和19个MT-CYB基因P/LP变异中的九个变异为无效变异。接近62%(16/26)的线粒体复合体IV的P/LP变异为无效变异(附表S2)。
mt-mRNA 线粒体信使RNA,LP 可能致病的,P 致病的 线粒体复合体I的变异容易导致Leigh综合征(LS),MELAS和LHON。这些变异在肌肉中的异质率通常高于80%。但是,某些变异的异质率可能较低。例如在肌肉中:m.12706T>C(p.F124L,MT-ND5)的异质率在43-50%时会引起LS[36.37];m.13513G> A的异质率在29-77%时会导致MELAS,在31-95%时会导致LS[38]。LHON变异通常在血液甚至所有组织中都是同质性的。而导致LS或MELAS相关的变异也可能在异质率低时引起LHON。例如,m.13094T> C(p.V253A,MT-ND5)在尿液中的异质性为40%时会导致LHON[39]。 MT-CYB变异通常会导致运动不耐受。有些也可能引起脑病;例如m.14787delTTAA(p.I14fs)、m.15092G> A(p.G116S)、m.15242G> A(p.G166 *)和m.15579A> G(p.Y278C)。这些变异异质性的阈值大约为80%。 线粒体复合体IV的缺陷通常会引起肌病(m.6698delA(p.K265fs271 *,MT-CO1))。然而,还可能出现其他症状,例如脑病(m.7023G> A(p.V374M,MT-CO1)),周围神经病变(m.7222A> G(p.Y440C,MT-CO1))以及发育迟缓和肌张力减退(m.6526T> C(p.M208T,MT-CO1))(附表S2)。在受累个体中,MT-CO1和MT-CO2基因的P/LP变异的异质水平通常较高。 总之,对mt-mRNA变异进行恰当的评估是精准的临床诊断和遗传咨询的基础。我们从以前的病例中推导出了一套变异分类标准,并使用我们的新病例来检验和完善这套标准,进而将其应用于解读已报导的mt-mRNA VUS变异和新变异。对mt-mRNA变异进行分类的最关键信息包括变异的类型,对患者及其母系亲属的不同组织的异质率定量分析,与异质率相关的功能研究,人群数据以及计算机模拟预测。对于LHON变异,LHON患者携带率高于正常对照组人群也是证明其致病性的重要线索。此外,当收集到更多世界上不同的单倍群基因背景和不同种群数据时,有助于更好地解读罕见变异。本研究是基于我们的实验室经验。为了更好地应用这些变异解读标准,不同实验室必须根据其检测和质量控制情况来验证这些标准。(来源遗传学办公室)返回搜狐,查看更多 |
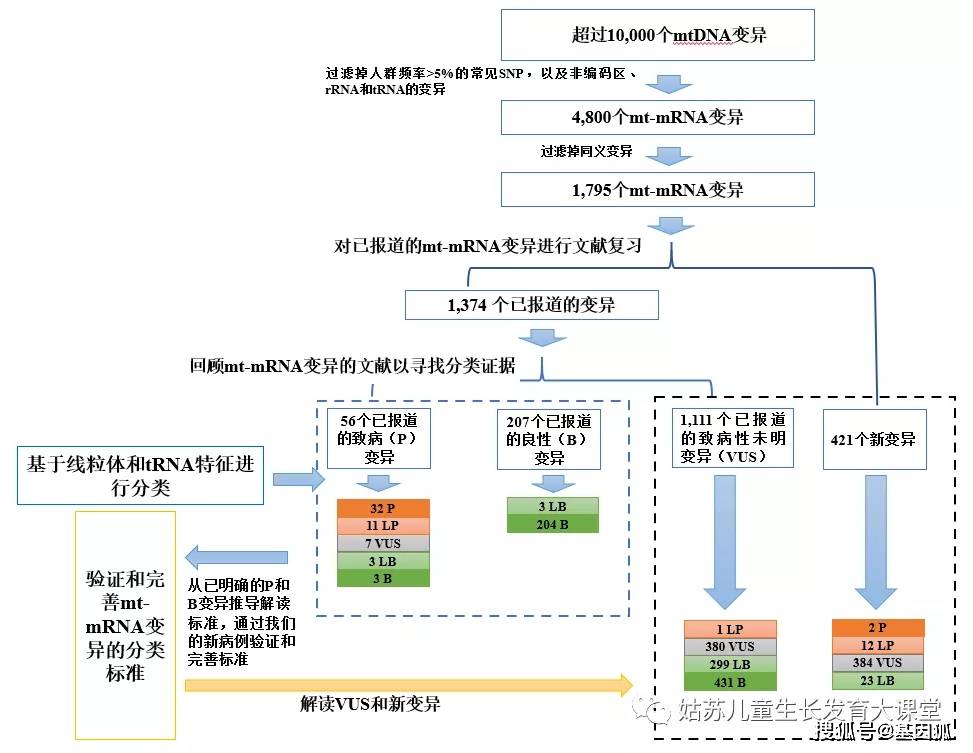
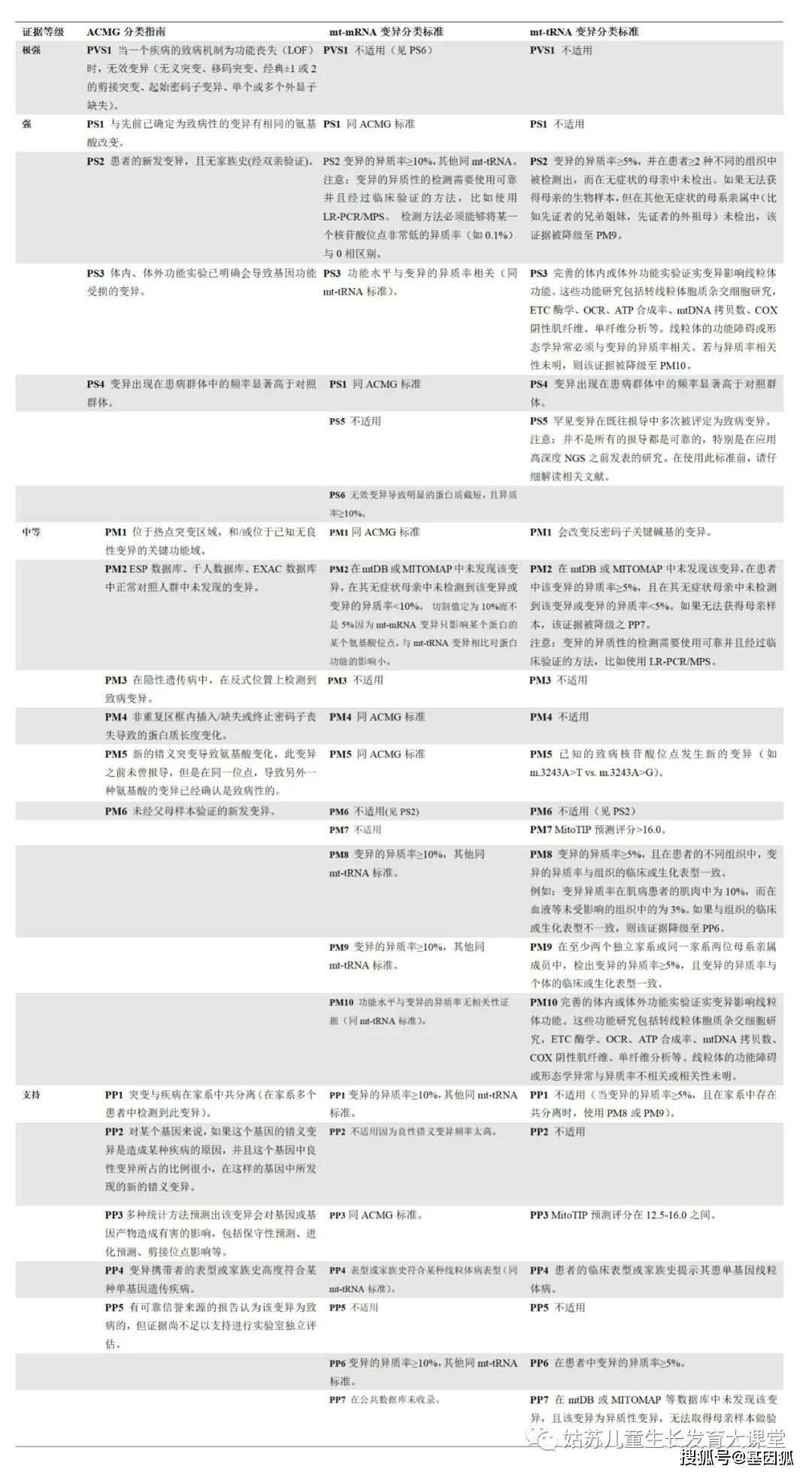 表1. mt-mRNA致病变异分类标准
表1. mt-mRNA致病变异分类标准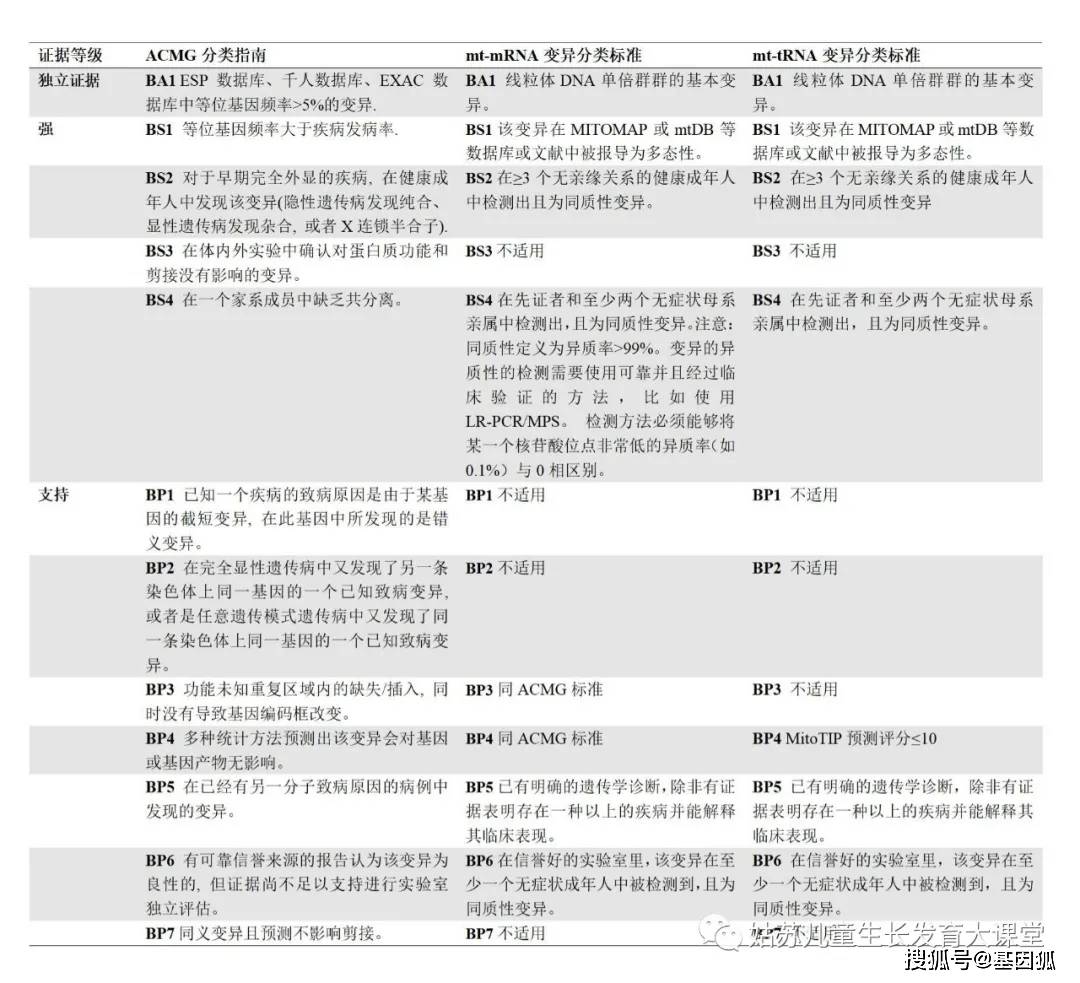 表3.原报导为致病变异但重新评估后分类不一致的mt-mRNA变异
表3.原报导为致病变异但重新评估后分类不一致的mt-mRNA变异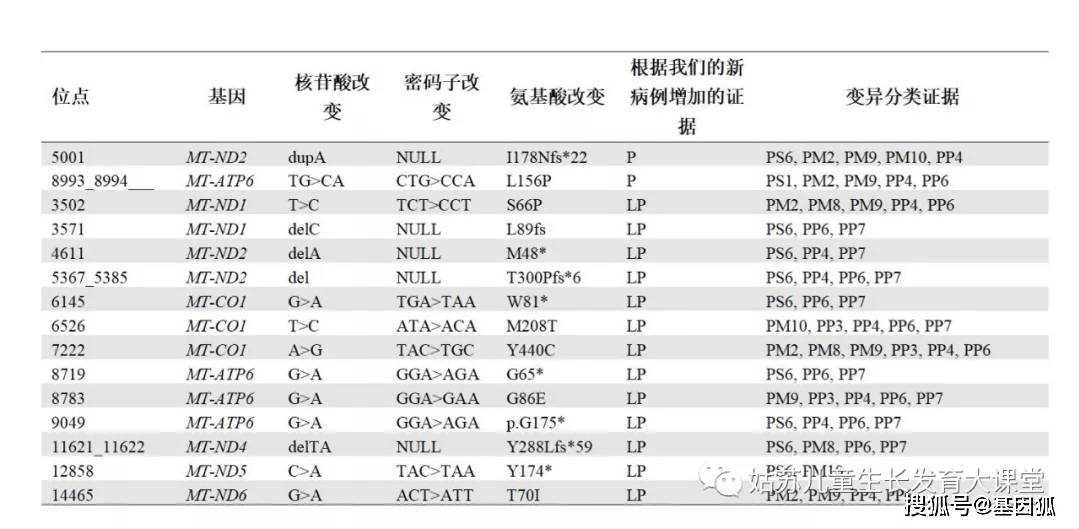 表4. 致病的或可能致病的新变异
表4. 致病的或可能致病的新变异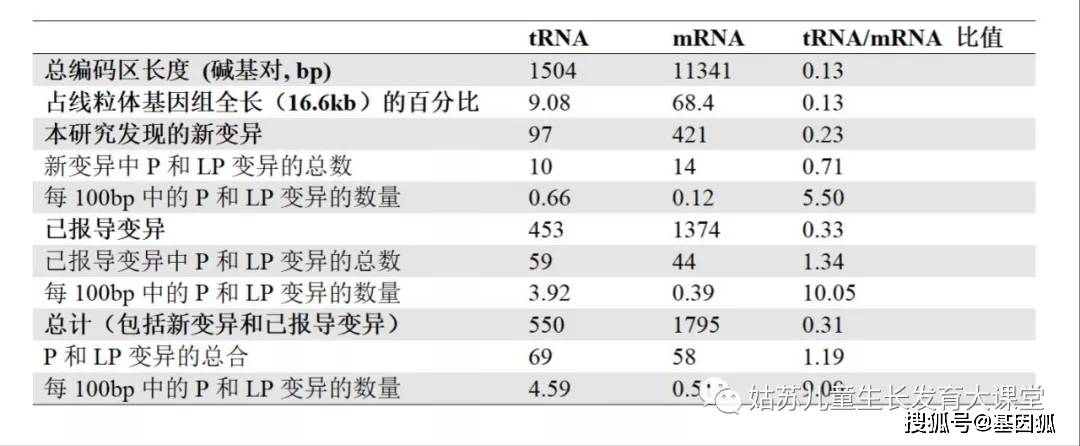 表5. mt-tRNA和mt-mRNA变异的分布
表5. mt-tRNA和mt-mRNA变异的分布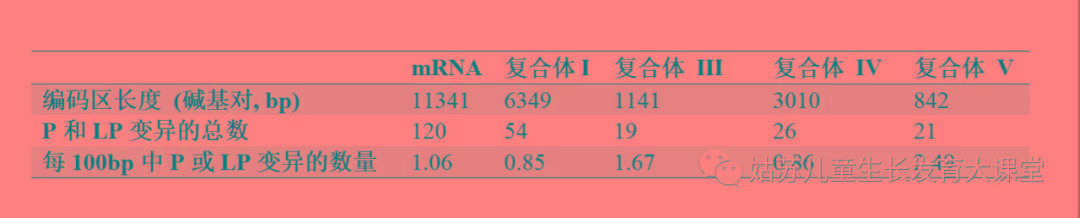 表6. 已报导的致病和可能致病的mt-mRNA变异(包括本研究报导和MITOMAP中收录的)在不同复合体之间的分布。
表6. 已报导的致病和可能致病的mt-mRNA变异(包括本研究报导和MITOMAP中收录的)在不同复合体之间的分布。【本文地址】
今日新闻 |
推荐新闻 |