一种特异性结合GPC3蛋白的纳米抗体GN1及其制备方法和应用与流程 |
您所在的位置:网站首页 › gpc3蛋白大小 › 一种特异性结合GPC3蛋白的纳米抗体GN1及其制备方法和应用与流程 |
一种特异性结合GPC3蛋白的纳米抗体GN1及其制备方法和应用与流程
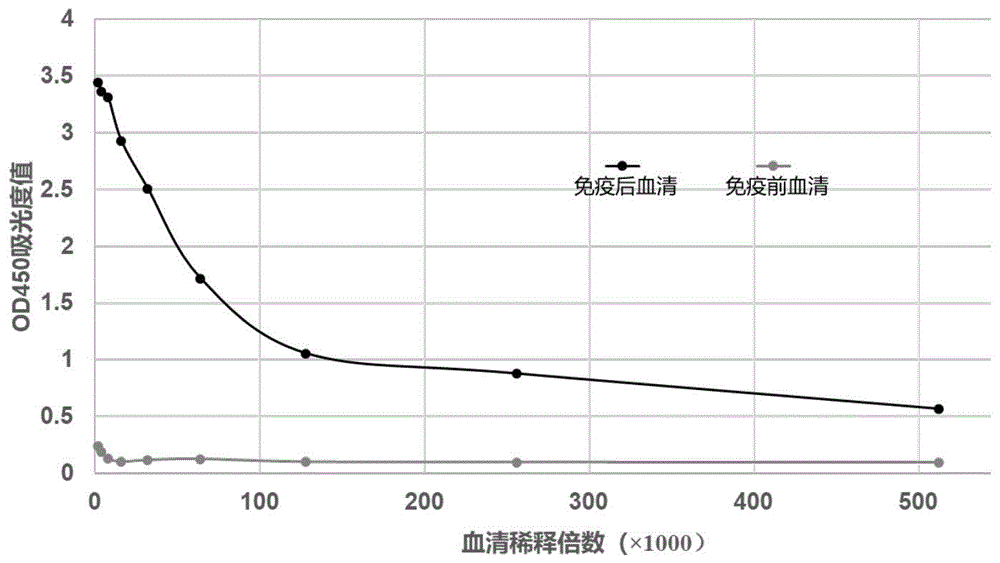 本发明涉及生物技术领域,具体涉及一种特异性结合gpc3蛋白的纳米抗体gn1及其制备方法和应用。 背景技术: 磷脂酰肌醇蛋白聚糖3(glypican-3,gpc3)是磷脂酰肌醇蛋白聚糖(glypican)家族中的一员,通过糖基磷脂酰肌醇(gpi)锚定附着在细胞表面。gpc3在肝癌细胞中异常高表达,但是在正常组织中表达有限,gpc3的高表达与肝癌的预后差呈正相关。此外,在肝癌患者血液中可检测出分泌型gpc3蛋白。因此,gpc3成为肝癌诊断治疗的新靶点。 目前,单克隆抗体在癌症的检测及靶向治疗方面成功的应用,引起了肿瘤治疗的变革。然而,传统的单抗(150kd)分子质量过大,肿瘤组织穿透能力差,造成肿瘤区域的有效浓度较低,治疗效果不充分,并且具有很高的免疫原性。改造的抗体很难达到原来的亲和力,阻碍了抗体检测的灵敏性。此外,完全人源化的传统抗体开发周期长,生产成本高,稳定性不够等诸多因素限制其在临床中的应用。 本领域需要安全有效的靶向结合gpc3用于诊断和治疗gpc3相关病症(如肝癌)的抗体试剂或抗体药物,及筛选该纳米抗体的方法。本发明满足了这一需求。 技术实现要素: 为了解决现有技术中的一个或者多个上述问题,本发明在第一方面提供了一种特异性结合gpc3蛋白的纳米抗体gn1,其中: 所述纳米抗体gn1由重链抗体的可变区组成; 所述重链抗体的可变区包括抗原决定簇互补区(complementarity-determiningregion,cdr)和骨架区(frameworkfrgion,fr); 所述骨架区选自由fr1、fr2、fr3和fr4以及与其具有不低于80%,优选不低于90%,更优选不低于95%,进一步优选不低于99%的同一性的氨基酸序列组成的组,所述抗原决定簇互补区选自由cdr1、cdr2和cdr3以及与其具有不低于80%,优选不低于90%,更优选不低于95%,进一步优选不低于99%的同一性的氨基酸序列组成的组; 所述cdr1的氨基酸序列具有如seqidno.1所示的序列,所述cdr2的氨基酸序列具有如seqidno.2所示的序列,所述cdr3的氨基酸序列具有如seqidno.3所示的序列;所述fr1的氨基酸序列具有如seqidno.4所示的序列;所述fr2的氨基酸序列具有如seqidno.5所示的序列;所述fr3的氨基酸序列具有如seqidno.6所示的序列;所述fr4的氨基酸序列具有如seqidno.7所述的序列。 在一些优选的实施方式中,所述纳米抗体gn1的氨基酸序列如序列表中seqidno.8所示。 本发明在第二方面提供了一种多核苷酸,所述多核苷酸编码本发明第一方面所述的纳米抗体gn1。 在一些优选的实施方式中,所述核苷酸序列如序列表中seqidno.9所示。 本发明在第三方面提供了一种重组载体,所述重组载体包含与调控序列可操作地连接的本发明第二方面所述的多核苷酸,或包含本发明第二方面所述的核苷酸序列中缺失1-5个氨基酸残基的密码子和/或进行1-5个碱基对的错义突变得到的核苷酸序列。 本发明在第四方面提供了一种宿主细胞,所述宿主细胞包含本发明第三方面所述的重组载体。 本发明在第五方面提供了一种药学组合物,所述药学组合物包含本发明第一方面所述的纳米抗体gn1和药学上可接受的载体,优选的是,所述药物组合物用于检测和/或治疗肝癌,更优选的是,所述肝癌为小肝癌。 本发明在第六方面提供了一种试剂盒,所述试剂盒包括本发明第一方面所述的纳米抗体gn1,优选的是,所述药物组合物用于检测和/或治疗肝癌,更优选的是,所述肝癌为小肝癌。 本发明在第七方面提供了一种用于制备本发明第一方面所述的纳米抗体gn1的方法,所述方法包括以下步骤: (1)免疫羊驼:使用gpc3蛋白对羊驼进行免疫; (2)提取rna:采用来自经免疫的羊驼的外周血来提取总rna; (3)纳米抗体基因库构建:采用所述总rna构建纳米抗体基因库; (4)淘选并鉴定:采用偶联链霉亲和素的磁珠和生物素化gpc3蛋白对纳米抗体基因库进行亲和淘选,并使用gpc3多克隆抗体通过夹心phageelisa鉴定阳性克隆。 (5)表达和纯化:挑选phageelisa信号最高的阳性克隆菌株诱导表达纳米抗体(命名gn1),并分离纯化得到纳米抗体gn1。 在一些优选的实施方式中,在步骤(1)中,所述gpc3蛋白为真核细胞hek293表达的蛋白;所述羊驼为成年健康羊驼;和免疫采用皮下多点注射进行。 在另一些优选的实施方式中,在步骤(2)中,采用来自经免疫的羊驼的外周血的淋巴细胞来提取总rna。 在另一些优选的实施方式中,在步骤(3)中,构建纳米抗体基因库包括如下步骤: a.cdna的合成:将所述总rna反转录合成cdna链; b.羊驼重链抗体可变区基因的扩增:以所述cdna链为模板,分别使用两对引物经pcr扩增获得羊驼重链抗体igg2和重链抗体igg3的可变区(vhh)基因,所述两对应物包括由hs引物和hanti1引物组成的第一对引物和由引物hs引物和hanti2引物组成的第二对引物; c.将vhh基因连接到载体上:将vhh基因连接到质粒载体中并纯化回收包含重组载体的连接产物;优选的是,所述连接采用基于同源重组原理的试剂盒例如clonexpressultraonestepcloningkit试剂盒(南京诺唯赞生物科技有限公司(vazymebiotechco.,ltd))进行,所述质粒载体为pcomb3x载体,并且所述纯化回收使用pcr产物纯化试剂盒进行; d.重组载体的转化:通过电击转化法将连接产物转化到大肠杆菌中并检测库容量;优选的是,所述大肠杆菌为er2738大肠杆菌;更优选的是,所述电击转化通过如下方式进行:对于er2738大肠杆菌和连接产物的混合物,使用tip头轻轻搅动2-3圈避免产生气泡,加入到冷却的1.0mm电转化杯中,轻微甩动电转杯使得混合物全部进入电击区域,立刻放置电转仪,电转条件:1400-1600v,200-400ω,10μf,3.5-4.5毫秒,立刻加入975μl预热的复苏培养基,上下吹动三次混匀细胞,转入细菌培养管中37℃,250rpm,复苏培养1h;进一步优选的是,所述检测取1μl菌液稀释10-1、10-3、10-5、10-7倍数后在含有氨苄青霉素的lb培养基的平板进行涂板; e.基因库进行噬菌体展示:将步骤d中复苏培养的转化菌转入200ml含氨苄青霉素和四环素的sb培养基中培养,37℃,250rpm培养至细菌对数期,加入滴度为1012pfu的m13ko7辅助噬菌体,37℃静置侵染30min,220-250rpm摇培1h后加卡纳霉素至终浓度70μg/ml并过夜摇培;次日,过夜菌以13000rpm的转速在4℃离心10min,取上清液并加入5×peg/nacl,冰浴2-4h后进行离心,获得的沉淀重悬在保护缓冲液(含有1×蛋白酶抑制剂,0.02%叠氮钠,0.5%bsa的pbs溶液)中,用0.22μm滤膜过滤后分装放置-80℃,即获得纳米抗体噬菌体基因库。 在另一些优选的实施方式中,在步骤(4)中,所述淘选重复3-4次;优选的是,所述鉴定通过如下方式进行:在淘选结束后,挑取克隆进行培养,使用gpc3多克隆抗体通过夹心phageelisa鉴定阳性克隆,提取elisa信号值最高的克隆质粒进行测序,获得纳米抗体gn1的核苷酸序列和纳米抗体gn1的氨基酸序列。 在另一些优选的实施方式中,在步骤(5)中,所述表达和纯化通过如下方式进行:利用所述阳性克隆对应的菌株提取质粒,将质粒转入大肠杆菌top10f'中,挑取单克隆培养后,进行诱导表达,收集菌体裂解细菌后,采用镍柱纯化,并经咪唑洗涤后获得抗gpc3蛋白的纳米抗体gn1。 在另一些优选的实施方式中,在步骤(3)中,所述载体为pcomb3x载体;pcr扩增使用的hs引物具有如seqidno.10所示的序列;所述hanti1引物具有如seqidno.11所示的序列;和/或所述hanti2引物具有如seqidno.12所示的序列即。 在另一些优选的实施方式中,在步骤(4)中,所述淘选包括如下子步骤: ①取200μl链霉素亲和磁珠,用1mltbst清洗两次后加入1ml封闭液,其中第一轮筛选用3%bsa封闭液,第二轮筛选用3%脱脂牛奶封闭液,并且3%bsa与3%脱脂牛奶交替使用,4℃、150-160rpm震荡封闭1h; ②将噬菌体文库加入相同的封闭液于4℃、150-160rpm震荡封闭除杂; ③封闭后的磁珠放置于磁力架30s,吸弃上清,加入1mltbst清洗3次; ④清洗后的磁珠加入200μl的结合缓冲液和30μl的生物素化的gpc3蛋白中,4℃、130-150rpm震荡结合30min,之后加入除杂的噬菌体文库,4℃、150-160rpm震荡结合1h,所述结合缓冲液包含20mmph7.5的tris-hcl、0.5mnacl和1mmedta; ⑤反应液置入磁力架30s,吸弃上清,加入1mltbst清洗8-12次后,磁珠中加入200μlph为2.2的甘氨酸-盐酸,并于150-200rpm下,震荡结合10-15min,置入磁力架30s,吸出上清立即加入1.6μlph为9.1的tris-hcl中和洗脱产物,获得第一次产物; ⑥取10μl第一次产物进行滴度测定,其余第一次产物进行扩增。洗脱产物扩增的方法:洗脱产物加入3-5ml对数期生长的er2738细菌,37℃静置孵育30-45分钟后,加入到5ml37℃预热的含有200μg氨苄青霉素和60μg四环素抗性的sb培养基中,37℃220-250rpm摇培1小时,补充加入500μg氨苄青霉素,37℃220-250rpm继续摇培1小时后,加入1012pfum13ko7辅助噬菌体,37℃静置侵染30min,转移至91ml预热的含有9.4mg氨苄青霉素和920μg四环素的sb培养基中,220-250rpm摇培1h后加卡纳霉素至终浓度70μg/ml并过夜摇培。次日,过夜菌以13000rpm的转速在4℃离心10min,取上清液并加入5×peg/nacl,冰浴2-4h后进行离心,获得的沉淀重悬在0.01mpbs缓冲液中,用于下一轮淘选。 ⑦第一次产物扩增液用于投入第二轮淘选;同理,第二轮产物扩增液投入第三轮筛选。第二轮和第三轮生物素化gpc3蛋白使用体积依次降低至6μl、1.5μl。 本发明在第八方面提供了本发明第一方面所述的纳米抗体gn1或本发明第二方面所述的多核苷酸或本发明第三方面所述的重组载体或本发明第四方面所述的宿主细胞在制备用于检测和/或治疗肝癌的药物或试剂盒中的应用。优选的是,所述检测选自由诊断试剂免疫检测、流式检测、细胞免疫荧光检测组成的组中的应用。 本发明具有如下技术效果: (1)分子量小,纳米抗体gn1是目前最小的抗体分子(仅15kda),其分子量是普通抗体的1/10。 (2)结构稳定,耐热性能好。纳米抗体gn1除了具备单克隆抗体的抗原结合性能外,在强变性剂或高温的条件下表现出不易变性稳定性强的特点,亲和力高。 (3)对人体的免疫原性弱; (4)肿瘤组织穿透力强; (5)比常规抗体更容易储藏和运输; (6)更易表达、基因工程化改造,作为诊断试剂或治疗抗体更具有优势。 (7)本发明方法采用真核细胞(hek293细胞)表达的gpc3蛋白进行免疫,hek293细胞真核细胞表达的蛋白比原核细胞表达的蛋白更接近蛋白的天然构象,保留了更多的表位,刺激羊驼产生高效价的抗体,保证了基因文库的多样性。采用多对引物进行重链抗体的重链可变区(vhh)基因扩增,提高扩增得到的vhh基因的多样性;采用缺失甲基转移酶的宿主扩增噬菌粒载体,可以制备高质量的载体,保证酶切效果。利用同源重组酶,增加连接效率,降低载体自连效率;采用液相淘选方法,能够暴露更多的表位,减少了固相筛选时因为抗原包埋而封闭一些表位的概率,实现更多表位抗体的全面筛选。在夹心phageelisa(夹心噬菌体酶联免疫吸附测定法)中,使用gpc3多克隆抗体取代gpc3蛋白包埋在elisa平板上筛选阳性克隆,能避免由于gpc3蛋白包埋直接吸附在板上时,一些疏水的表位吸附在板上,从而降低了噬菌体文库与其结合的概率,造成阳性克隆丢失。 纳米抗体gn1应用于制备诊断和治疗肝癌的药物,编码纳米抗体gn1的基因或含有其基因的质粒或含有基因的重组质粒或含有的其基因的重组质粒的重组细胞应用于制备免疫检测诊断试剂、流式检测、细胞免疫荧光检测和治疗肝癌的药物中的应用。 附图说明 图1是羊驼(vicugnapacos)免疫血清效价检测的图。横坐标是血清稀释倍数,纵坐标是相应的吸光度od450值。根据阳性血清与阴性血清之比(positive/negetive)≥2.1为阳性,可见免疫后羊驼血清效价可达到1:512000。说明免疫效果很好,羊驼体内产生了大量抗体。 图2是gn1纳米抗体热稳定性实验结果图。图2显示,纳米抗体结构稳定,90℃处理2h后仍然保持活性,但80℃处理2h后gpc3-mab已经失去活性。说明与gpc3-mab相比,本发明的纳米抗体结构稳定,耐热性能好。 图3是vhh基因扩增产物。图中泳道1是引物hs和hanti1的扩增产物igg3的vhh基因,泳道2是引物hs和hanti2的扩增产物igg2的vhh基因。泳道3是分子量标准物。 图4是pcomb3x噬菌粒载体sfiⅰ酶切电泳图。图4的泳道1是pcomb3x噬菌粒载体转化大肠杆菌dh5α扩增后,提取质粒进行sfiⅰ酶切结果。因为质粒被甲基化,所以sfiⅰ酶没有切开质粒。图4的泳道2是pcomb3x噬菌粒载体转化大肠杆菌c2925扩增后提取质粒进行sfiⅰ酶切结果。大肠杆菌c2925是甲基化酶缺陷细菌,pcomb3x质粒没有被甲基化,所以sfiⅰ酶切成功。泳道m表示分子量标准物。 图5是pcomb3x噬菌粒载体和vhh基因连接效率的检测。a.使用同源重组酶,增加了载体和vhh基因的连接效率,随机挑选的28个克隆都是阳性克隆,载体自连效率为0%。b.使用t4dna连接酶,随机挑选28个克隆,10个是没有连接vhh基因的载体,载体自连效率为35%。 图6采用梯度稀释法评估文库的库容。a.使用同源重组酶连接pcomb3x载体和vhh基因构建文库的库容;取10μl文库分别稀释成10-4、10-5、10-6涂板后的克隆数目,10-6平板克隆数30个,所以vhh基因文库库容为3.0×109cfu/ml。b.使用t4连接酶连接pcomb3x载体和vhh基因构建文库的库容;取10μl文库分别稀释成10-3、10-4、10-5涂板后的克隆数目,10-5平板克隆数29个,所以vhh基因文库库容为2.9×108cfu/ml。由此可知,同源重组酶连接vhh基因和载体构建的基因文库库容比使用t4dna连接酶连接vhh基因和载体构建的基因文库提高十倍。 图7为gpc3纳米抗体gn1的sds-page蛋白质电泳图,图中泳道m代表分子量标记物(marker),泳道1:纳米抗体gn1。gpc3纳米抗体gn1的分子量大小约为15kda。 图8为纳米抗体gn1与肝癌细胞株hepg2(gcp3阳性)的结合图,激光共聚焦结果显示纳米抗体gn1结合在hepg2细胞株的细胞膜上。说明纳米抗体gn1能够特异性结合gpc3阳性细胞,即纳米抗体gn1特异性结合gpc3蛋白。 图9为流式细胞术检测gn1与肝癌靶细胞hepg2结合率。a.阴性对照组(blank空白对照);b.肝癌细胞阳性组(gn1纳米抗体)。 图10为gn2-luc融合蛋白sds-page电泳图。图中泳道m:分子量标准物;泳道1:200mm咪唑洗脱蛋白;泳道2:100mm咪唑洗脱蛋白;泳道3:50mm咪唑洗脱蛋白;泳道4:细胞裂解液流经镍柱的流穿液;泳道5:诱导表达后细菌菌液。结果发现,gn2-luc融合蛋白分子量大小约为35kda。 图11为融合蛋白gn1-luc为基础的夹心elisa方法的建立。图a为荧光强度曲线,显示gpc3蛋白最佳检测范围51.9ng/ml-157.4ng/ml(即图中ec20-ec80范围之间,其中ec20、ec80相当于荧光强度分别为最强荧光20%、80%时gpc3蛋白含量)。肝癌患者血清gpc3含量范围:150ng/ml-300ng/ml(gastroenterology2003;125:89–97)。因此该检测方法适用于肝癌的诊断。图b为该方法示意图。 图12为gn1纳米抗体抑制gpc3阳性肝癌细胞的增殖。图中可见gn1纳米抗体能够抑制gpc3阳性肝癌细胞hep3b、hepg2、huh7的增殖,抑制效率分别高达59.99%、66.24%、70.04%。并且gn1对gpc3阴性肝癌细胞sk-hep1、bel7404的增殖没有抑制作用。说明gn1纳米抗体能够抑制gpc3阳性肝癌细胞的增殖,可以作为抗肝癌抗体药物应用于肝癌的治疗。 具体实施方式 以下实施例用于说明本发明,但不用来限制本发明的范围。在不背离本发明精神和实质的情况下,对本发明方法、步骤或条件所作的修改或替换,均属于本发明的范围。 实施例1:纳米抗体gn1的制备 纳米抗体gn1的制备过程包括以下步骤: (1)免疫羊驼: 取1mg真核细胞hek293细胞表达的gpc3蛋白与弗氏完全佐剂乳化,共计2ml,采用皮下多点注射的方式对一只健康成年羊驼进行第一次疫;第15天,0.5mggpc3蛋白与弗氏完全佐剂乳化,共计2ml,皮下多点注射进行第二次免疫;之后每隔7天采用0.5mggpc3蛋白与弗氏不完全佐剂乳化获得共计2ml的免疫用乳化注射液进行下一次免疫。共免疫6次,每次免疫后第7天采血检测效价。检测血清效价后采集外周血100ml。发明人发现,使用hek293细胞真核细胞表达的蛋白比原核细胞表达的蛋白更接近蛋白的天然构象,保留了更多的表位,刺激羊驼产生高效价的抗体(图1),保证了基因文库的多样性。 (2)100ml外周血用来提取淋巴细胞提取rna: 使用leukolocktm(thermofisher)按照说明书操作步骤提取总rna。 (3)纳米抗体基因库的构建: 纳米抗体基因库的构建包括如下步骤: a.合成cdna链:参照iiifirst-strandsynthesissystemforrt-pcr说明书将8μgrna反转录合成cdna链; b.扩增羊驼vhh基因:以步骤a的cdna链为模板,使用引物hs和hanti1进行4次pcr反应,扩增羊驼重链抗体igg3的vhh基因。使用引物hs和hanti2进行6次pcr反应扩增羊驼重链抗体igg2的vhh基因(图3)。本发明人发现,vhh基因来自于重链抗体igg2和重链抗体igg3,所以用这两对引物扩增的vhh基因既包括igg2的vhh基因,又包括igg3的vhh基因,相对于其他研究机构使用的vhh-p1和vhh-p2(只扩增igg2的vhh基因)构建的基因库多样性更加丰富。 反应体系如下:cdna2μl;hs引物1.3μl;hanti1引物或者hanti2引物1.3μl;taqenzymemix44.4μl;反应程序:94℃,3min;94℃,30s,55℃,30s,72℃,1min,32个循环;72℃,5min。pcr产物通过电泳进行分析,并且切胶回收pcr产物(约500bp单一条带)。 c.pcomb3x噬菌粒载体sfiⅰ酶切线性化处理:本发明人发现,因为sfiⅰ酶对甲基化的dna敏感,所以该载体扩增时的宿主菌选择大肠杆菌c2925,大肠杆菌c2925缺失非特异性核酸内切酶i(enda1)活性,缺失甲基转移酶,nna序列不会被甲基化,可用于扩增高质量pcomb3x质粒,并且不影响sfiⅰ的酶切效果。于是,购买大肠杆菌c2925感受态细胞,pcomb3x噬菌粒载体转化大肠杆菌c2925感受态细胞。取1μlpcomb3x噬菌粒载体加入融化的大肠杆菌c2925感受态细胞中,冰水中静置10-20min,42℃热激60-90s,冰浴2min,加入25-37℃的soc培养基,37℃,250rpm,培养1h。取200μl菌液涂氨苄青霉素抗性平板,放置37℃过夜培养。次日挑选单克隆至氨苄青霉素抗性lb培养基37℃,250rpm过夜培养,使用质粒提取试剂盒提取质粒。在0.2μlpcr管中进行酶切反应:取1μg质粒pcomb3x,4μlsfiⅰ酶,4μlcutsmartbuffer,加超纯水去离子水补充至50μl。放置pcr仪设置50℃酶切12h(图4)。次日,取酶切产物电泳,切胶回收大片段,溶解在超纯去离子水。 d.根据同源重组原理,把vhh基因连接到pcomb3x载体上:使用clonexpressultraonestepcloningkit试剂盒,pcomb3x载体大片段(sfiⅰ酶切后切胶回收)200ng,vhh基因60ng,2×clonexpressmix5μl,超纯水去离子水补充至10μl,放置50℃,反应5分钟后立即放置冰上冷却。使用pcr产物纯化试剂盒对连接产物进行纯化回收。本发明人发现,由于利用同源重组酶,因此增加了载体和vhh基因的连接效率,并且由于整个体系无dna连接酶,因此降低了载体自连的效率(图5),提高了库容量(图6)。 e.重组载体的电击转化:将步骤d纯化的连接产物3μl加入到50μl大肠杆菌er2738电转化感受态细胞中,使用tip头轻轻搅动1-2圈避免产生气泡,电转体系加入到冷却的1.0mm电转化杯中,手腕轻轻甩动电转杯使得细胞沉入电转杯底部,立刻放到电转仪,电转条件:1400-1600v,200-400ω,10μf,3.5-4.5毫秒,立刻加入975μl预热的复苏培养基,上下吹动三次混匀细胞,转入细菌培养管中37℃,250rpm,复苏培养1h;取1μl菌液稀释10-1、10-3、10-5、10-7倍数后涂板(氨苄青霉素抗性的lb)检测库容量。从平板随机挑取28个克隆进行菌落pcr验证连接效率(图5)。菌落pcr的引物为:引物hs和引物hback(seqidno.13)。 f.基因库救援:将步骤e复苏培养的转化菌转入200ml含氨苄青霉素和四环素的s培养基中培养,37℃,250rpm培养至细菌对数期,加入1012pfum13ko7辅助噬菌体37℃静置侵染30min,摇培1h后加卡纳霉素(终浓度70μg/ml)过夜摇培。次日,过夜菌离心(13000rpm,4℃)10min,取上清液并加入5×peg/nacl,冰浴2h后进行离心,获得的沉淀重悬在保护缓冲液(含有1×蛋白酶抑制剂,0.02%叠氮钠,0.5%bsa的pbs溶液),用0.22μm滤膜过滤后分装放置-80℃,即获得纳米抗体噬菌体基因库。 (4)淘选gpc3纳米抗体gn1并鉴定:对步骤f获得的噬菌体文库采用偶联链霉亲和素的磁珠进行亲和淘选,获得第一次产物(output),取10μl产物测滴度,其余产物进行扩增后进行第二轮和第三轮筛选,再从第三轮筛选得到的培养平板中挑取96个克隆,37℃过夜培养,通过phageelisa鉴定阳性克隆。本发明人发现,与包被抗原蛋白的固相筛选方法相比,磁珠液相筛选能够暴露更多的表位,减少了固相筛选时因为抗原包埋而封闭一些表位的概率,实现更多表位抗体的全面筛选。 上述亲和淘选的方法如下: a、取200μl链霉素亲和磁珠,用1mltbst清洗两次后加入1ml封闭液(第一轮筛选用3%bsa,第二轮筛选用3%脱脂牛奶,3%bsa与3%脱脂牛奶交替使用),4℃、150-160rpm震荡封闭1h; b、将噬菌体文库加入相同的封闭液于4℃、150-160rpm震荡封闭除杂; c、封闭后的磁珠低速(2000-3000rpm)离心30s,吸弃上清,加入1mltbst清洗3次; d、清洗后的磁珠加入200μl的磁珠结合缓冲液(20mmph7.5tris-hcl,0.5mnacl,1mmedta)和30μl的生物素化的gpc3蛋白中,4℃、130-150rpm震荡结合30min,之后加入除杂的噬菌体文库,4℃、150-160rpm震荡结合1h; e、反应液低速(2000-3000rpm)离心30s,吸弃上清,加入1mltbst清洗10次,磁珠中加入200μlph为2.2的甘氨酸-盐酸,并于150-200rpm下,震荡结合15min,低速(2000-3000rpm)离心30s,吸出上清立即加入1.6μlph为9.1的tris-hcl中和洗脱产物,即获得第一次产物(output); f、取10μl步骤e获得饿产物测滴度,其余产物进行扩增,扩增方法如下:洗脱产物加入3-5ml对数期生长的er2738细菌,37℃静置孵育30-45分钟后,加入到5ml37℃预热的含有200μg氨苄青霉素和60μg四环素抗性的sb培养基中,37℃220-250rpm摇培1小时,补充加入500μg氨苄青霉素,37℃220-250rpm继续摇培1小时后,加入1012pfum13ko7辅助噬菌体,37℃静置侵染30min,转移至91ml预热的含有9.4mg氨苄青霉素和920μg四环素的sb培养基中,220-250rpm摇培1h后加卡纳霉素至终浓度70μg/ml并过夜摇培。次日,过夜菌以13000rpm的转速在4℃离心10min,取上清液并加入5×peg/nacl,冰浴2-4h后进行离心,获得的沉淀重悬在0.01mpbs缓冲液中,用于下一轮淘选。 g、第一次产物扩增液用于投入第二轮淘选;同理,第二轮产物扩增液投入第三轮筛选。第二轮和第三轮生物素化gpc3蛋白使用体积依次降低至6μl、1.5μl。 h、从三轮筛选的平板上挑选46个克隆做夹心phageelisa鉴定阳性克隆。使用商品化的gpc3多抗(bio-techne生物公司)用包被缓冲液稀释成5μg/ml,每孔100μl,加入96孔板,4℃过夜包被。第二天洗板后,加入gpc3蛋白,37℃孵育1-2h;洗板后每孔加入300μl5%脱脂牛奶封闭液封闭1h。洗板拍干后放置4℃备用。使用灭菌牙签挑取46个单克隆,接种于96深孔培养板,每孔800μl氨苄抗性培养基,37℃,250rpm震荡培养约5-6h,菌液od600约0.6-0.8,每孔加入1011pfum13ko7侵染,37℃静置30min,37℃,250rpm震荡培养1h,加入卡纳霉素至终浓度70μg/ml,37℃,250rpm过夜培养。次日深空板6000rpm离心15min,取上清加入3%脱脂牛奶除杂后,加入到放置4℃备用的elisa96孔板中,室温150-160rpm震荡结合1h。洗板机洗3次,加入抗m13的二抗,室温150-160rpm震荡结合30min,加入羊抗兔hrp抗体150-160rpm震荡结合30min后,tmb显色分析。 (5)gpc3纳米抗体gn1表达和纯化:将步骤(4)获得的elisa信号最强阳性菌株,采用质粒试剂盒(qiagen公司提供)提取质粒,用于转化大肠杆菌top10f',挑取单克隆37℃过夜培养后;按照1:100比例,加入至100mlsb培养基,37℃培养3-4h至od600达到0.6-0.8;加入iptg(终浓度0.5mmol/l),26℃过夜诱导表达;第二天上午离心(10000rpm,10min)收集菌体,使用细菌蛋白提取试剂(b-per)室温裂解30min,裂解细菌释放蛋白,再次离心(12000rpm,15min),收集上清加入镍柱,4℃结合3-6h,使用20mmol/m的咪唑洗涤4个柱体积,分别收集5ml50mmol/l、100mmol/l咪唑洗涤液,即获得纳米抗体gn1(图7)。 挑选步骤(4)获得的阳性克隆,用于测序,获得纳米抗体gn1的核苷酸序列(如序列表中seqidno.9所示),进而根据密码子表获得纳米抗体gn1的氨基酸序列(如序列表中seqidno.1所示)。 实施例2:纳米抗体gn1热稳定性实验: (1)包被gpc3蛋白,1μg/mlgpc3蛋白,100μl每孔加入酶标板中,4℃过夜包被。 (2)pbst洗板三次后,每孔加入300μl5%脱脂牛奶,37℃封闭1小时。 (3)每孔分别加入4℃,37℃,60℃,70℃,80℃,90℃环境下处理2小时的本发明纳米抗体gn1和商品化gpc3单克隆抗体(gpc3-mab,invitrogen公司)。每孔100μl,室温孵育结合1小时后,pbst洗板3次。 (4)gn1组,因为gn1抗体有ha标签,所以每孔加入hrp酶标记的ha-mab(santacruz公司),室温孵育40分钟,pbst洗板3次,加入tmb显色10分钟,用2m硫酸终止反应后,酶标仪检测450nm处紫外吸光度(od450值)(参见图2)。 (5)gpc3-mab组,加入hrp酶标记抗鼠igg二抗(santacruz公司),室温孵育30分钟后,pbst洗板3次,加入tmb显色10分钟,用2m硫酸终止反应后,酶标仪检测450nm处紫外吸光度(od450值)(参见图2)。 实施例3:以gn1纳米抗体为基础的gn1-荧光素酶融合蛋白检测血清gpc3蛋白的夹心elisa方法的建立: (1)扩增gn1基因。以实施例1步骤(4)获得的质粒为模板,通过pcr扩增gn1核苷酸序列;pcr产物使用ncoi和sfii进行酶切,pcr产物纯化试剂盒对酶切产物进行纯化回收。 (2)构建融合基因gn1-luc。合成荧光报告基因即纳米荧光素酶基因(nano-luciferase,简称luc)(荧光蛋白报告基因荧光素酶的氨基酸序列如seqidno.14所示):亚克隆至载体pet22b的notⅰ和salⅰ位点之间,ncoi和sfii双酶切含有荧光报告基因纳米荧光素酶的载体pet22b,切胶回收包含荧光素酶基因的pet22b载体骨架序列;步骤(2)回收的载体骨架序列和步骤(1)纯化的pcr产物,在t4连接酶作用下连接构建融合基因gn1-luc,连接产物转化大肠杆菌bl21(de3)感受态细胞,挑选测序正确的阳性克隆,进行诱导表达。 (3)融合蛋白gn1-luc的表达与纯化。挑选阳性克隆,在4ml含有氨苄青霉素抗性的sb培养基中,37℃、220rpm,过夜培养。第二天取过夜培养菌按照1:100接种至200ml含有氨苄青霉素抗性的sb培养基中,37℃、220rpm培养至od600达到0.6左右,加入iptg(终浓度0.5mm,220rpm,过夜诱导表达融合蛋白gn1-luc。第二天上午离心(10000rpm,10min)收集菌体,使用细菌蛋白提取试剂(b-per)室温裂解30min,裂解细菌释放蛋白,再次离心(12000rpm,15min),收集上清加入镍柱,4℃结合3-6h,使用20mmol/m和40mmol/m的咪唑洗涤4个柱体积,收集5ml100mmol/l咪唑洗涤液,即获得融合蛋白gn1-luc(图10),发明人发现纳米抗体结构简单,更易于基因工程化操作构建融合蛋白,本发明中使用原核表达即获得融合蛋白gn1-luc。 (4)以融合蛋白gn1-luc为基础的夹心elisa方法的建立。商品化的gpc3多抗(bio-techne生物公司)用包被缓冲液稀释成3μg/ml,每孔100μl,加入96孔板,4℃过夜包被。第二天,以pbst为洗脱液,使用洗板机洗3次后加入300μl5%的脱脂牛奶封闭1h。洗板机洗3次,加入不同浓度的gpc3蛋白,室温150-160rpm震荡结合1h。洗板机洗3次,加入0.2μg/ml的融合蛋白gn1-luci,室温150-160rpm震荡结合0.5h,洗板机洗5次,拍干96孔板后,每孔加入100μl荧光素酶底物腔肠素(coelenterazine)-h(5μg/ml),使用酶标仪立即检测荧光信号(图11)。 实施例4:流式细胞术检测纳米抗体gn1与靶细胞hepg2的结合效率 该实验选择高表达gpc3的hepg2细胞作为靶细胞。收集1×105-106hepg2细胞重悬在500μlpbs(2%bsa),加入1μggn1纳米抗体4℃孵育结合30min与高表达gpc3的靶细胞hepg2细胞孵育结合,150-160rpm缓慢震动以防止非特异性结合。pbs洗涤细胞后加入ha-tag(c29f4)rabbitmab(peconjugate)histagpercp-conjugatedantibody,4℃孵育630min,pbs清洗细胞3次后重悬在500μlpbs缓冲液,流式细胞术检测gn1纳米抗体与hepg2细胞的结合效率(图9)。 实施例5:荧光成像观察纳米抗体gn1对gpc3阳性肝癌细胞的靶向结合: 共聚焦扫描荧光显微镜观察纳米抗体gn1与gpc3性肝癌细胞的特异性结合,具体方法为:将纳米抗体gn1(1μg)加入1-5×106个hepg2肝癌细胞(gpc3阳性)中,于4℃下避光孵育20-40min,pbs洗涤2次后,加入5μl带有pe抗-ha标签的抗体(peanti-hatagantibody)(abcam,clone:20a12)于4℃下孵育20-40min,用pbs(磷酸缓冲盐溶液)洗涤2次后,将样品置于共聚焦扫描荧光显微镜观察纳米抗体gn1与hepg2肝癌细胞的特异性结合(图8)。 实施例6:纳米抗体gn1对gpc3阳性肝癌细胞增殖的抑制: 24孔板培养细胞,控制每孔细胞个数大约2-3×104时加入纳米抗体gn1(20μm),同时使用人免疫球蛋白higg作为对照;肝癌细胞系选择gpc3阳性肝癌细胞(hep3b、hepg2和huh-7细胞)和gpc3阴性肝癌细胞(sk-hep1和bel-7404)。培养4-5天后,每孔加入mtt溶液(5mg/ml)继续孵育4h后,小心吸取并弃掉上清液体,每孔加入150μldmso,振荡10min使结晶充分溶解,使用酶标仪检测490nm波长(图12)。 表1.实施例中扩增得到的产物或所采用的引物的序列。 最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。 sequencelisting 广西科技大学 一种特异性结合gpc3蛋白的纳米抗体gn1及其制备方法和应用 gy19100390 14 patentinversion3.5 1 8 prt vicugnapacos 1 glyargthrphevalglyserthr 15 2 9 prt vicugnapacos 2 sermetsertrpleuglyargserthr 15 3 17 prt vicugnapacos 3 alaleualaleuglytyrileaspleualaargproaspalavalasp 151015 ala 4 25 prt vicugnapacos 4 glnvalglnleuvalgluserglyglyglyleuvalglnalaglygly 151015 serleuargleusercysalaalaser 2025 5 17 prt vicugnapacos 5 metglytrppheargglnalaproglylysgluarggluphevalala 151015 arg 6 38 prt vicugnapacos 6 tyrtyralaaspservallysglyargphethrileserargaspasn 151015 alalysasnthrvaltyrleuglnmetasnserleulysprogluasp 202530 thralavaltyrtyrcys 35 7 11 prt vicugnapacos 7 trpglyglnglythrleuvalthrvalserser 1510 8 124 prt vicugnapacos 8 glnvalglnleuvalgluserglyglyglyleuvalglnalaglygly 151015 serleuargleusercysalaalaserglyargthrphevalglyser 202530 thrmetglytrppheargglnalaproglylysgluargglupheval 354045 alaargmetsertrpleuglyargserthrtyrtyralaaspserval 505560 lysglyargphethrileserargaspasnalalysasnthrvaltyr 65707580 leuglnmetasnserleulysprogluaspthralavaltyrtyrcys 859095 alaleualaleuglytyrileaspleualaargproaspalavalasp 100105110 alatrpglyglnglythrleuvalthrvalserser 115120 9 372 dna vicugnapacos 9 caggtgcagctcgtggagtctgggggaggattggtgcaggctgggggctctctcagactc60 tcctgtgcagcctctgggcgcaccttcgttggctcaaccatgggctggttccgccaggct120 ccagggaaggagcgtgagtttgtagcacggatgagctggcttggtcgtagcacatactat180 gcagactccgtgaagggccgattcaccatctccagagacaacgccaagaacacggtttat240 ctgcaaatgaacagcctgaaacctgaggacacggccgtttattactgtgcactcgcactt300 ggttatatcgaccttgcacgtccggacgcagtggacgcatggggccaggggaccctggtc360 actgtctcctca372 10 52 dna 人工序列 10 tggctggtttcgctaccgtggcccaggcggcccagktgcagctcgtggagtc52 11 41 dna 人工序列 11 tggtgatggtgctggccgatgggggtcttcgctgtggtgcg41 12 42 dna 人工序列 12 tggtgatggtgctggccggtcttgtggttttggtgtcttggg42 13 24 dna 人工序列 13 gcccccttattagcgtttgccatc24 14 180 prt 人工序列 14 argvalphethrleugluaspphevalglyasptrpargglnthrala 151015 glytyrasnleuaspglnvalleugluglnglyglyvalserserleu 202530 pheglnasnleuglyvalservalthrproileglnargilevalleu 354045 serglygluasnglyleulysileaspilehisvalileileprotyr 505560 gluglyleuserglyaspglnmetglyglnileglulysilephelys 65707580 valvaltyrprovalaspasphishisphelysvalileleuhistyr 859095 glythrleuvalileaspglyvalthrproasnmetileasptyrphe 100105110 glyargprotyrgluglyilealavalpheaspglylyslysilethr 115120125 valthrglythrleutrpasnglyasnlysileileaspgluargleu 130135140 ileasnproaspglyserleuleupheargvalthrileasnglyval 145150155160 thrglytrpargleucysgluargileleualavalasplysleuala 165170175 alaalaleuglu 180 |
【本文地址】