一种验证CRISPR |
您所在的位置:网站首页 › ctc密码子 › 一种验证CRISPR |
一种验证CRISPR
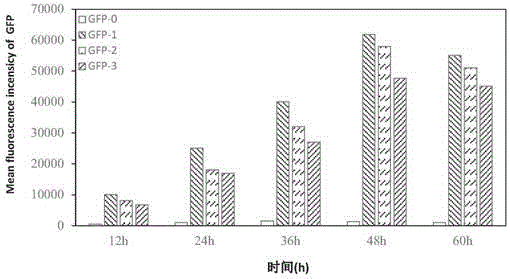 本发明涉及基因插入领域,更具体地,涉及一种验证crispr-cas9系统介导目标基因插入产朊假丝酵母的可行性的方法。 背景技术: 酵母是简单的单细胞真核生物,它具有严密的真核基因表达调控系统,还具有种类丰富、易培养、大多无致病性等特点。酿酒酵母和毕赤酵母已被作为细胞工厂用来生产各种外源蛋白,但其作为宿主仍存在多处不足,比如大量的宿主酵母会出现质粒缺失的现象;酿酒酵母通常在发酵过程中会有乙醇产生,不利于高密度发酵;重组蛋白在酵母中表达时通常会发生超糖基化的现象;毕赤酵母在表达外源蛋白时需要添加甲醇进行诱导,这些缺陷限制了酵母表达系统的发展,因此,需要开发一种安全、高效的酵母表达系统用于食品、药物蛋白的生产。 产朊假丝酵母(candidautilis),是可以作为食品添加剂直接添加使用的安全酵母,可以将其运用于食品、制药等行业。产朊假丝酵母也是工业生产中非常重要的一种微生物,目前已作为宿主用来生产氨基酸、单细胞蛋白饲料和谷胱甘肽等。与毕赤酵母和酿酒酵母相比,产朊假丝酵母具有以下特性:1.可以同时利用五碳糖和六碳糖;2.易培养,能以硝酸盐和尿素作为氮源,能以糖蜜作为营养物质进行生长,且培养基中不需要加入任何的生长因子;3.在严格好氧培养条件下不具有酵解抑制有氧氧化效应,因此不会生成副产物乙醇;4.可以作为食品添加剂直接添加使用。基于上述产朊假丝酵母的优点,建立一种产朊假丝酵母表达系统对酵母表达系统的工业应用、发展具有必要性。 由于产朊假丝酵母中没有稳定的附加型质粒,现有的产朊假丝酵母表达系统主要采用整合型表达载体将外源基因整合到产朊假丝酵母染色体上;但c.utilis基因组是多倍体菌株,利用整合型载体进行同源重组时,由于所有的染色体不能同时整合成功,整合片段会容易发生回复突变,即整合进入产朊假丝酵母基因组中的目的片段会被重新从基因组中释放出来;而且外源基因整合时一般都带有抗性筛选标记基因,该筛选标记基因较难去除,导致整个整合过程繁琐复杂。而利用游离型的载体表达外源基因则会存在载体稳定性的问题,为维持载体的稳定,需要在培养基里添加抗生素,增加了成本也带来抗生素滥用等问题。c.utilis另外一种常规的的基因改造方法是采用cre-loxp重组酶系统,这个系统存在操作繁琐、耗时长等缺点。 因此,现有技术中缺乏针对于产朊假丝酵母简单而有效的基因编辑方法,对于产朊假丝酵母工程菌株的改造来说是个技术瓶颈。此外,针对于产朊假丝酵母的基因编辑方法还有zfns方法、talens技术,但zfns方法中模块组装的锌指蛋白活性比较低,有效的锌指蛋白并不容易获得,该方法的成本较高,talens技术则需要组装多个重复模块,过程非常繁琐。现有技术中还存在一种利用crispr-cas9系统的基因改造方法,以下简称cas9系统,与前两种基因编辑技术相比,crispr-cas9系统中的载体构建更简单、方便,且应用该系统的基因编辑方法在编辑上具有非常高的精确性,其应用领域也更广阔。作为一种新型的基因编辑技术crispr-cas9可通过单一的引导rna(sgrna)引导cas9蛋白识别目标序列并切断dna双链,使细胞以同源重组或非同源末端连接的方式修复受损dna,从而实现对目标基因的靶向敲除、插入等目的。 若cas9系统能够应用于产朊假丝酵母的基因插入操作中,则有助于产朊假丝酵母新表达系统的实现,结合产朊假丝酵母自身特点,提供一种新型的产朊假丝酵母基因插入方法能够为外源基因在产朊假丝酵母中表达提供新途径,为产朊假丝酵母的广泛应用提供基础,但现有技术中cas9系统能否应用于产朊假丝酵母的基因插入操作仍未被确认。 技术实现要素: 本发明旨在克服上述现有技术的至少一种缺陷,提供一种验证crispr-cas9系统介导目标基因插入产朊假丝酵母的可行性的方法,利用crispr-cas9系统实施目标基因的插入,并对插入状况进行鉴定,以验证crispr-cas9系统介导目标基因插入产朊假丝酵母基因组是否可行,同时,验证的过程中实现sgrna重组质粒、cas9产朊假丝酵母表达载体、左右同源臂重组供体片段的构建。 本发明采取的技术方案是,一种验证crispr-cas9系统介导目标基因插入产朊假丝酵母的可行性的方法,包括以下步骤: s1、使用限制性核酸内切酶对酵母cas9表达载体和第一产朊假丝酵母游离型表达载体进行双酶切,并连接产生的cas9片段和第一游离型表达载体骨架片段,获得重组质粒pcggαal-cas9;提取酵母cas9表达载体质粒和第一产朊假丝酵母游离型表达载体质粒后,对两个质粒进行双酶切,双酶切则保证了cas9片段与第一游离型表达载体骨架片段的导向性连接,避免基因片段连接方向出错,减小了后续筛选过程的工作量。对酶切片段进行琼脂凝胶电泳并胶回收包含cas9编码区的片段与第一游离型表达载体骨架片段。将回收到的不含启动子的cas9表达盒片段和含有启动子的第一游离型表达载体片段进行连接,优选的,cas9表达盒与游离型表达载体骨架按照摩尔比10∶1的比例进行连接。 优选的,连接产物还需进行阳性转化子的鉴定,所述阳性转化子鉴定采用pcr鉴定方式。进行阳性转化子鉴定后,挑选单株阳性转化子,提取并获得重组质粒pcggαal-cas9,即将cas9片段转入至在产朊假丝酵母中可自主复制的游离型表达载体中。 优选的,获得重组质粒后还需进行阳性克隆筛选、cas9基因表达检测、质粒拷贝数鉴定,以挑选能够转化成功的质粒并确认转化后的重组质粒pcggαal-cas9能在产朊假丝酵母中正常表达;优选的,所述阳性克隆筛选利用游离型表达载体所携带的抗性基因进行培养筛选,经多代培养后提取多代后克隆体的酵母基因组进行pcr扩增鉴定;优选的,cas9基因表达检测采用rt-pcr方式检测;优选的,通过相对定量法,以产朊假丝酵母中的个别酶基因作为对照来测定cas9基因在产朊假丝酵母中的拷贝数。 s2、扩增产朊假丝酵母编码谷氨酸的转运rna即trnaglu基因、sgrna下游表达单元,所述sgrna下游表达单元以crrna为模板进行扩增,并分别引入iis类限制性核酸内切酶酶切位点,trnaglu基因与sgrna下游表达单元扩增产物通过酶切位点进行重叠pcr连接;重叠后的片段以及第二产朊假丝酵母游离型表达载体经双酶切后连接在一起,获得载体pglu-sgrna。根据产朊假丝酵母ctc密码子的trna序列,以产朊假丝酵母基因组为模板扩增得到trnaglu片段,将trnaglu片段作为加工后的sgrna转录启动子,有助于提高基因编辑效率。成熟sgrna全长96bp,由20bp靶位点识别序列以及76bp的sgrna结构区构成,如果想更换一个靶位点,仅仅只需要更改这20bp的识别序列,而iis类限制性核酸内切酶具有切割位点在识别位点之外的特点,利用iis类限制性核酸内切酶能够不受酶切位点的限制而根据需要插入目标序列;以bsmbi为例,bsmbi为iis类限制性核酸内切酶,识别cgtctcnnnnn,切割位点为最后一个c下游的第二个碱基,产生4个碱基的5’突出粘性末端,因此可以不受酶切位点的限制而根据需要插入目标序列,利用bsmbi的特点,如图1所示,在sgrna表达载体上引入一段包含两个连续但方向相反的bsmbi位点的序列,酶切后可以产生两个不同的5’突出粘性末端,而且不会有酶切位点识别序列残留。如此,在构建新的sgrna表达载体时,只需要在20bp的识别序列,两个5’端加上与bsmbi酶切后产生的粘性末端的互补序列,即可方便的将识别序列连接到表达载体上,从而构建出针对新靶位点的sgrna载体。相对于其他iis类限制性核酸内切酶同样可以采取上述方式,进行新识别序列的构造。 优选的,扩增trnaglu基因片段、sgrna下游表达片段时,引入iis类限制性核酸内切酶酶切位点,方便构造新识别序列,同时利用该iis类限制性核酸内切酶酶切位点可进行trnaglu基因片段、sgrna下游表达片段的重叠pcr连接,构造能够插入识别位点的加工后sgrna。 优选的,本发明以trna作为sgrna的转录启动子;根据文献expressionofcrispr/cassingleguidernasusingsmalltrnapromoters可知,依靠trna转录后加工过程,产生成熟的sgrna可以获得比常用的snr52启动子更高的基因编辑效率。根据产朊假丝酵母的密码子使用频率,本发明选择了谷氨酸(glu)ctc密码子的trna作为sgrna的启动序列。除此之外,采用其他表达水平较高的转运rna基因也可进行验证。 优选的,s2具体包括以下步骤: (1)扩增产朊假丝酵母编码谷氨酸的转运rna(trnaglu)基因,扩增过程中的下游引物5’端引入iis类限制性核酸内切酶酶切位点;扩增sgrna下游表达单元,扩增过程中的上游引物5’端引入方向相反的两个iis类限制性核酸内切酶酶切位点;所述sgrna下游表达单元包括sgrna的结构区以及转录终止子区,sgrna扩增过程中,上游引物单链状态下5’端引入的一端序列在引物产生互补链成为双链时形成方向相反的两个iis类限制性核酸内切酶酶切位点。 (2)将扩增的trnaglu基因与sgrna扩增产物进行重叠pcr连接获得连接产物,分别对连接产物和第二产朊假丝酵母游离型表达载体进行双酶切,并连接酶切后的连接产物、游离型表达载体骨架片段,获得载体pglu-sgrna。扩增后的trnaglu基因与sgrna扩增片段通过相同方向的iis类限制性核酸内切酶酶切位点进行重叠pcr连接,以结合trnaglu基因片段和sgrna基因片段,并提供方向相反的iis类限制性核酸内切酶酶切位点以供靶点识别序列替换或插入。获得重叠pcr片段后,提取第二产朊假丝酵母游离型表达载体质粒,对该质粒、重叠pcr片段进行双酶切获得sgrna表达盒和游离型表达载体骨架。将所述sgrna表达盒和第二游离型表达载体骨架进行连接;所述双酶切位点并不与iis类限制性核酸内切酶酶切位点交叉,双酶切采用的具体酶与所述具体的iis类限制性核酸内切酶不同。优选的,sgrna表达盒片段与第二游离型表达载体骨架按照摩尔比10∶1的比例进行连接。 优选的,获得sgrna表达盒和第二游离型表达载体骨架连接后需进行阳性转化子的鉴定,进行阳性转化子鉴定后获得载体pglu-sgrna。 上述第一、第二产朊假丝酵母游离型表达载体具备双酶切位点,能够在产朊假丝酵母中进行自主复制。所述酵母cas9表达载体具备双酶切位点,cas9编码区处于双酶切位点之间,且cas9编码区两端的酶切位点应满足酶切连接后cas9启动子下游单元表达方向与第一产朊假丝酵母游离型表达载体表达方向一致。同理,sgrna表达盒与第二产朊假丝酵母表达载体基因上下游表达方向一致。 优选的,步骤s1中双酶切采用的酶为限制性内切酶spei和kpni;步骤s2双酶切采用的酶为限制性内切酶psti和bamhi。 s3、选定产朊假丝酵母基因中的预插入序列和靶点序列,所述靶点序列为预插入序列的一部分,所述靶点序列长20bp;sgrna具有20bp的识别序列区,采用20bp的靶点序列有助于构造识别靶点序列的sgrna,sgrna通过靶点序列能够识别带有靶点序列的预插入序列,并结合cas9系统对预插入序列基因片段进行切割,从而使得供体片段能够通过同源重组方式插入至预插入序列中。结合上述构造sgrna表达盒的过程,分别在靶点序列及靶点序列的互补序列的5’端加上iis类限制性核酸内切酶产生的粘性末端,经退火形成带有5’突出粘性末端的双链片段,将双链片段与iis类限制性核酸内切酶酶切载体pglu-sgrna后获得的骨架连接,获得重组质粒pglu-sgrna-预插入序列,即sgrna重组质粒;由于靶点序列加上的iis类限制性核酸内切酶产生的粘性末端能够该双链片段能够与pglu-sgrna中iis类限制性核酸内切酶酶切产生的末端匹配,从而将靶点序列插入至原pglu-sgrna重组质粒中,该质粒表达产生携带有靶点识别序列的成熟sgrna,有助于识别产朊假丝酵母基因组中包含靶点序列的预插入序列。 优选的,上述iis类限制性核酸内切酶为bsmbi酶。 优选的,所述预插入序列为pgk启动子序列,所选的靶点序列为: agcaggcctacaattacggatgg,下划线为pam序列; 所述靶点序列如序列表中的seqidno.1序列所示,所述pgk启动子序列如序列表中的seqidno.2所示。 s4、构建含有预插入序列左右同源臂的重组供体片段;在sgrna识别到预插入序列后,cas9系统进行切割,预插入序列基因片段为了修复断开的基因片段则会产生同源重组修复,在此过程中提供包含预插入序列左右同源臂的重组供体片段,能够让修复过程以供体片段为模板,进而实现重组供体片段的插入。 优选的,s4具体包括以下步骤: 所述步骤s4中的同源重组供体片段构建的步骤包括: (1)设计上下游引物扩增供体片段; (2)设计上下游引物扩增预插入序列,并在预插入序列上游引物5’端引入供体片段下游引物的反向互补序列; (3)通过重叠pcr获得包含预插入序列同源臂的重组供体片段。 除上述步骤外,针对于不同结构的供体片段或预插入序列片段可相应改变构建的方向和方式。 优选的,所述供体片段为gfp基因片段或果胶甲酯酶基因片段,供体片段不同则需要相应的检测、鉴定基因表达方式,即验证方式也存在不同。除此之外,采用其他基因片段也能用于验证cas9系统在产朊假丝酵母上的应用是否可行。 s5、sgrna重组质粒和携带重组供体片段的表达载体转化携带重组质粒pcggαal-cas9的产朊假丝酵母,直接或间接测定供体片段的基因表达水平验证cas9介导目标基因插入产朊假丝酵母是否可行。成熟sgrna与重组供体片段得到表达,并配合产朊假丝酵母内表达的cas9系统进行基因插入,所述供体片段插入成功则会在基因表达产物和/或基因序列和/或受基因插入的产朊假丝酵母表型中得以体现,通过测定供体片段的基因表达水平能够验证基因插入是否成功,进而验证通过cas9介导目标基因插入产朊假丝酵母的可行性。优选的,对预插入基因的选定和/或供体片段做替换实现多组对照实验,实现较为准确的验证结果。 优选的,基于上述sgrna重组质粒和携带重组供体片段的表达载体转化携带重组质粒pcggαal-cas9的产朊假丝酵母的前提下,还可将sgrna重组质粒和携带重组供体片段的表达载体转化至野生型产朊假丝酵母中,通过比较携带重组质粒pcggαal-cas9的产朊假丝酵母和野生型产朊假丝酵母之间供体片段的表达差异进一步验证供体片段是否成功插入,进一步验证cas9系统介导目标基因插入产朊假丝酵母的可行性。 优选的,上述重叠pcr产物或重组质粒或重组产生的载体均需进行阳性克隆筛选,避免未连接或未转化成功的产物影响最终的基因插入结果。 优选的,上述步骤中实际操作过程可能会涉及到大肠杆菌质粒提取、琼脂糖凝胶电泳、pcr或酶切产物回收纯化、凝胶回收dna、制备大肠杆菌感受态细胞、大肠杆菌的转化、制备产朊假丝酵母感受态细胞、产朊假丝酵母的电转化、酵母基因组的提取、总rna的提取、t载克隆等操作,该类操作均采用生物领域的通用方法,此类通用方法及步骤具体如下所示: (一)大肠杆菌质粒的提取 收集5ml过夜培养的菌液,参照天根生化科技有限公司的tianprepminiplasmidkit试剂盒提取质粒,具体步骤如下:(1)10,000rpm离心2min,倒弃上清液,收集菌体。(2)加入300μlbufferp1/rnasea混合液,彻底重悬细胞。(3)加入300μlbufferp2,轻轻混匀8次使菌体充分裂解,静置1min。(4)加入400μlbufferp3,立即轻轻上下颠倒8次,充分混匀。10,000rpm离心5min。(5)将上一步的上清液倒入收集管吸附柱中,10,000rpm离心1min。(6)倒弃滤液,将吸附柱放入收集管中。向吸附柱中加入600μlbufferpd,10,000rpm离心1min。(7)倒弃滤液,将吸附柱重新放回收集管中,向吸附柱中加入600μlbufferpw(已经用无水乙醇稀释),10,000rpm离心1min。(8)倒弃滤液,将吸附柱放入收集管中,10,000rpm离心2min,室温放置8min晾干柱子。(9)将吸附柱放置于干净的1.5ml离心管中,向吸附膜的中间部位滴加80μl50℃预热的h2o,室温放置3min,10,000rpm离心3min,丢弃柱子,用微量分光光度计测量其浓度后,质粒保存于-20℃。 (二)pcr产物或者酶切产物的琼脂糖凝胶电泳分析 参照《分子克隆实验指南(第三版)》,具体操作如下:(1)称取0.25g琼脂糖,加入25ml1×tae缓冲液后,使用微波炉加热使其溶解,冷却至55℃左右时,加入2μleb替代染料。混匀后将琼脂糖溶液倒入制胶板中,插入合适的梳子,室温下静置直至胶完全凝固。(2)取出梳子,将琼脂糖凝胶转移至电泳槽中,倒入1×tae电泳缓冲液至没过凝胶。依次将5μldnamaker和混有上样缓冲液的pcr产物加入凝胶孔中。(3)在120v恒定电压下电泳15min左右后,将凝胶转移至凝胶成像仪中,紫外灯下观察结果并保存。 (三)pcr产物或者酶促反应液中dna片段的回收纯化 根据凝胶dna微量回收试剂盒hipuregelmicrokit进行pcr产物或酶切产物的纯化回收,具体步骤如下:(1)短暂离心pcr产物或酶促反应液,用移液枪测量其体积后转移至1.5ml的离心管中。(2)加入等体积的buffergdp,涡旋混匀。(3)将吸附柱子套在收集管中,并将上一步的混合液转至dna吸附柱中。12,000×g离心1min,倒弃滤液;(4)加入300μlbuffergdp至柱子中,静置2min。12,000×g离心1min,倒弃滤液;(5)加入600μlbufferdw2至柱子中。12,000×g离心1min,倒弃滤液;(6)把柱子放回收集管中。12,000×g离心3min;(7)把柱子套在1.5ml离心管中,加入25μl的50℃预热的无菌水至柱子膜中央,放置3min。12,000×g离心2min,丢弃柱子,用微量分光光度计测其浓度后,把dna片段保存于-20℃。 (四)凝胶回收dna片段 具体步骤:(1)琼脂糖凝胶电泳结束后,将凝胶放置于紫外灯下,然后迅速切下含有目的dna片段的凝胶,并尽量去除多余的凝胶;(2)将切下的凝胶块转移至1.5ml的离心管中,称取凝胶的重量。按1mg凝胶块1μl体积计算,加入等体积的buffergdp。50℃水浴20min,期间上下颠倒离心管数次,让凝胶块溶解完全;(3)短暂离心收集液体后,将dna吸附柱套在2ml收集管中。把≤700μl溶胶液转移至柱子中。12,000×g离心2min;接下来的步骤按照上述方法(三)中的(4)~(8)步进行操作。 (五)大肠感受态的制备 大肠杆菌e.colidh5感受态细胞的制备,参照《分子克隆实验指南(第三版)》[50],具体操作如下:(1)取大肠杆菌dh5甘油保存菌以1%的接种量接种于5mllb液体培养基中,37℃,185rpm摇床培养过夜;(2)取过夜培养的的大肠杆菌dh5以1%的接种量转接至50mllb液体培养基中,37℃,185rpm培养,当培养液的od600在0.3-0.4时停止培养;(3)将菌液转移至无菌的50ml离心管中,冰浴15min后,于4℃,6,000×g离心5min;(4)倒弃上清液,加入30ml预冷的0.1mcacl2溶液,轻轻重悬菌体;(5)冰浴30min后,于4℃,8,000×g离心2min;(6)用移液枪小心吸尽上清液,加入3-5ml预冷的含15%甘油0.1mcacl2溶液轻轻重悬菌体,每管分装50μl,置于冰上待用,或于-80℃保存。 (六)大肠杆菌的转化 具体步骤:(1)取10μl连接产物或者质粒加入到50μl新鲜制备的大肠杆菌感受态中,轻轻吸打混匀,于冰上静置30min;(2)于42℃水浴热激90s后,冰上静置5min;(3)加入500μllb液体培养基,37℃,175rpm复苏45min;(4)8,000×g离心3min后,倒弃大部分上清液,剩余约200μl用来重悬菌体;(5)将重悬液全部涂于含相应抗生素的lb固体培养基上,于37℃恒温培养箱中倒置培养12h左右。 (七)产朊假丝酵母感受态的制备 具体步骤:(1)产朊假丝酵母的活化:将产朊假丝酵母甘油菌以2%接种量接种于5mlypd培养基中,30℃,200rpm摇床培养24h;(2)将活化后的产朊假丝酵母再次以2%的接种量接种于5mlypd培养基中,培养12h左右,转接至100mlypd培养基中,使其初始od600为1.0。30℃,200rpm摇床继续培养3h左右。稀释10倍后测菌液的od600在0.3~0.5时,停止培养,开始制备感受态细胞;(3)将菌液转移至无菌的50ml离心管中,冰上放置40min后,4℃,5,000×g离心5min,倒弃培养基,往离心管中加入8ml无菌水轻轻重悬菌体;(4)加入1ml10×tebuffer,1ml1m醋酸锂,轻轻混匀后于30℃,100rpm孵育50min;(5)之后加入300μl1mdtt轻轻混匀,30℃,185rpm孵育15min;(6)加预冷的无菌水至50ml,4℃,4,000×g离心8min,倒弃上清;(7)加入5ml预冷的1m山梨醇溶液,轻轻吸打重悬菌体,4℃,4,000×g离心8min;(8)倒弃上清,加入200μl预冷的1m山梨醇重悬菌体,使细胞终浓度约为1×1010个/ml;(9)将产朊假丝酵母感受态细胞以50μl/管分装到预冷的1.5ml无菌的离心管中,置于冰上待用。 (八)产朊假丝酵母的电转化 具体步骤:(1)向每管产朊假丝酵母感受态细胞中分别加入20μl(约5μg)的重组质粒,轻轻混匀,冰浴15min后转移至预冷的电转杯中;(2)电压为1.8kv,电击5ms后,立即加入1ml预冷的1m山梨醇溶液并混匀,常温静置15min;(3)在无菌条件下将电转杯中的菌液全部转移至无菌的4ml离心管中,加入2mlypd液体培养基并混匀,于30℃,175rpm复苏4h;(4)4,000×g离心8min后,倒弃大部分上清液,剩余200μl重悬菌体;(5)将菌体重悬液全部涂于含g418的ypd固体培养基上,30℃培养箱中倒置培养3-5天。 (九)酵母基因组的提取 根据美基生物公司的solpurednakits操作步骤:(1)吸取1ml酵母培养液至1.5ml离心管,12,000×g离心2min收集菌体;(2)加入300μlbufferse,20μllyticase(5u/μl)和20μl(1m)dtt至酵母细胞中。用移液枪重悬酵母细胞后,37℃水浴1h;(3)12,000×g离心2min收集酵母原生质体,小心弃去上清液;(4)加入400μlbufferwlb和2μlrnasesolution至原生质体中,高速涡旋30s后,65℃水浴45min;冰上放置3min后,加入100μlbufferpps至裂解液中,涡旋30s;(5)12,000×g离心5min,收集上清液倒入新的1.5ml离心管中;(6)加入400μl异丙醇至上清液中;(7)颠倒离心管30次,12,000×g离心1min;(8)小心倒弃上清液,并将离心管反扣于吸水纸上吸干剩余的溶液;(9)加入500μl70%乙醇至dna沉淀中,涡旋混匀,12,000×g离心1min;(10)小心倒弃上清液,把离心管反扣于吸水纸上,空气干燥15min;(11)加入200μl无菌水至dna沉淀中,涡旋5s;(12)65℃水浴45min溶解dna后,于-20℃保存。 (十)总rna的提取 产朊假丝酵母总rna的提取,参考博日科技有限公司的biozol总rna提取试剂操作步骤:(1)挑选培养至对数生长期的酵母细胞,将50ml酵母培养液转移至50ml的离心管中,4℃,12,000g离心3min收集菌体;(2)吸取菌体至研钵中,在液氮条件下将样品研磨成粉末;(3)在样本中加入1mlbiozol后,将匀浆样品在室温下孵育15min,使样品充分裂解至匀浆液呈透明;(4)将匀浆液4℃,12,000g离心5min后,弃沉淀取上清;(5)按照1∶5(氯仿∶biozol)的比例(v/v)加入200μl氯仿。盖紧管盖,涡旋30s混匀,将其置于冰上孵育15min后,于4℃,12,000×g离心10min后取上清。(6)将上清液转移至干净的1.5ml离心管中,加入等体积冰浴的异丙醇,上下颠倒10次混匀,将混匀的样品在-20℃条件下孵育40min,4℃,12,000×g离心10min。(7)弃上清,加入1ml冰预冷的75%的乙醇洗涤rna沉淀一次,颠倒洗涤离心管管壁,尽可能让沉淀悬浮起来,4℃,12,000×g离心10min,再次去除上清后,在阴凉处适度干燥rna沉淀。(避免过分干燥,那样会降低rna的可溶性)(8)用50μl的无rnase水溶解rna。抽提好的rna,可保存于-70℃。 (十一)t载克隆 (1)pcr产物加a:由于primestarhsdnapolymerase反应的产物是平末端,无法连接到t-vector上,因此需要加a反应,反应体系如下: 72℃反应15min后按照pcr产物纯化步骤纯化dna,并用微量分光光度计测定dna浓度,-20℃保存。 (2)按照thermofisherdnaligase试剂盒说明书,纯化的dna片段与pmd19-t载体连接,反应体系如下: 将连接产物按照上述方法(六)转化大肠杆菌。 除上述通用方法之外也可根据根据量级或菌株的不同做适应性调整,也可采用类似原理的通用方法实现操作。 与现有技术相比,本发明的有益效果为:应用crispr-cas9系统至产朊假丝酵母的基因插入,通过鉴定插入的供体片段基因表达状况确认是否成功将目标基因插入至预插入序列中,验证crispr-cas9系统介导目标基因插入至产朊假丝酵母基因组的可行性;初步验证crispr-cas9系统引导外源基因靶向插入产朊假丝酵母的基因组,为产朊假丝酵母基因工程的改造提供了新策略;同时,提供了应用crispr-cas9系统插入目标菌种的具体方法,为高效、快速、稳定地进行工业酵母菌株的定向基因改造奠定基础,也为其它微生物细胞的基因修饰研究提供了借鉴,具体方法还包括含cas9基因片段的产朊假丝酵母表达载体构建、sgrna重组质粒构建以及靶点识别序列插入sgrna、左右同源重组供体片段构建;根据产朊假丝酵母的基因表达状况,选择性的将trnaglu片段作为转录启动子,提高了产朊假丝酵母中基因编辑的效率;在sgrna重组质粒过程中引入iis类限制性核酸内切酶酶切位点,实现trnaglu和sgrna下游表达单元扩增产物的重叠pcr连接,并利用iis类限制性核酸内切酶酶切位点的特殊性在sgrna下游表达单元扩增过程中引入方向相反的iis类限制性核酸内切酶酶切位点,提供靶点识别序列插入点的同时不干扰trnaglu与其重叠pcr连接,实现了高效、简单的sgrna加工。通过sgrna重组质粒、携带重组供体片段的表达载体、携带cas9表达载体的产朊假丝酵母实现整个crispr-cas9系统的应用,当插入过程中发生插入失败或异常现象出现时,能够及时的判断整个系统哪个部分出错,并能针对于单个重组质粒或表达载体进行鉴定、重构,避免了整体的重构,减少了验证过程的时间,且单独构建质粒降低了插入过程的操作难度,在产朊假丝酵母能够通过crispr-cas9系统插入基因的前提下,提供了一种简单、高效的产朊假丝酵母基因组插入操作。此外,采用crispr-cas9基因编辑技术,只需先构建表达cas9的产朊假丝酵母,然后将针对特定位点的sgrna载体和外源基因的dna片段共转化进表达了cas9蛋白的产朊假丝酵母细胞,仅需一次转化,就可以实现高效外源基因定点插入基因组。由于同源重组片段并没有携带抗性基因,因此可以通过简单的无选择压力培养,即可将抗性基因去除,减少了繁琐的操作。 附图说明 图1为bsmbi酶切位点示意图; 图2为产朊假丝酵母游离型表达载体pcggαal-cas9构建流程图; 图3为含有pcggαal-cas9的大肠杆菌菌落pcr鉴定图; 图4为pcggαal-cas9转产朊假丝酵母阳性克隆的鉴定图; 图5为cas9基因转录表达pcr鉴定图; 图6为trnaglu启动子克隆电泳图; 图7为含有pglu-sgrna的大肠杆菌菌落pcr鉴定图; 图8为含pglu-sgrna-pgk的大肠杆菌菌落pcr鉴定图; 图9为供体片段pgk-gfp构建图; 图10为含pmd19t-pgk-gfp的转大肠杆菌菌落pcr鉴定图; 图11为供体片段pgk-gfp克隆电泳图; 图12为pglu-sgrna-pgk和供体片段pgk-gfp转产朊假丝酵母阳性克隆的鉴定图; 图13为荧光显微镜观察gfp的表达图; 图14为流式细胞仪检测gfp在重组酵母中的表达图; 图15为gfp阳性重组菌株筛选标记的去除点种平板图; 图16为流式细胞仪检测gfp在产朊假丝酵母中的表达; 图17为含pcpgαal-pmea的大肠杆菌菌落pcr鉴定图; 图18为pmea同源重组片段pgk-pmea的结构示意图; 图19为含pmd19-pgk-pmea的大肠杆菌的菌落pcr鉴定; 图20为pgk-pmea片段转产朊假丝酵母阳性克隆的筛选; 图21为果胶甲酯酶的pmea的sds-page分析图; 图22为pmea-3重组菌株去除抗性筛选标记结果; 图23为重组产朊假丝酵母cu-pgk-pmea的生长曲线和酶活曲线; 图24为产朊假丝酵母游离型表达载体pcuars-gapgαa结构图; 图25为产朊假丝酵母游离型表达载体pcuars-gapzαam结构图; 图26为产朊假丝酵母游离型表达载体pcuars-pgkg-gfp结构图; 图27为带有pmea基因的质粒pcggαal-pmea结构图。 具体实施方式 以下内容为本发明的进一步说明,不应理解为对本发明的限制。在没有违背本发明所涉及的技术方法实质下,对本发明的中的方法、条件和步骤进行简单的改动均属于本发明要求保护的范围。 实施例1 crispr-cas9介导报告基因gfp靶向整合及表达 本实施例所用的产朊假丝酵母atcc22023购于广东省微生物菌种保藏中心;酵母cas9表达载体p415-gall-hcas9和sgrna表达载体p426-crrna由addgene公司提供;产朊假丝酵母游离型表达载体:pcuars-gapgαa、pcuars-gapzαam,其中pcuars-gapgαa的结构和特殊序列如图24所示,pcuars-gapzαam的结构和特殊序列如图25所示;大肠杆菌克隆载体pmd19-t购自大连宝生物工程有限公司;引物合成及dna测序由invitrogen公司完成;引物序列-f代表上游引物,引物序列-r代表下游引物。 一、产朊假丝酵母cas9表达载体的构建与验证 1.提取质粒p415-gall-hcas9和pcuars-pgapgαa,然后用限制性内切酶spei和kpni分别酶切p415-gall-hcas9质粒和pcuars-gapgαa质粒,反应体系为50μl: 37℃水浴1h后,琼脂糖凝胶电泳分析酶切结果并保存,胶回收含cas9编码区的片段与质粒pcuars-gapgαa骨架片段。 2.cas9表达盒与质粒pcuars-gapgαa片段的连接 将回收到的不含启动子的cas9表达盒片段和含有gap启动子的载体pcuars-gapgαa骨架按照摩尔比10∶1的比例进行连接,连接体系为20μl: 22℃连接1h后,连接产物用来转化大肠杆菌dh5α,具体流程如图2所示。 3.阳性转化子的鉴定 待平板上长出转化子后,用无菌的牙签挑取少量菌体混于预先配置好的20μlpcr反应体系中进行pcr鉴定。本实施例中将菌株接种至含卡那霉素的lb平板,待含卡那霉素的lb平板长出单菌落即转化子,用无菌的牙签随机挑选5个单菌落用cas9-f和cas9-r进行菌落pcr鉴定,将菌体混于预先配置好的20μlpcr反应体系。其中,阴性对照不含模板。反应体系为: 然后分装,每管20μl。反应条件为: cas9引物如表1所示: 表1 pcr鉴定结果如图3所示,其中m:dnamakerdl2000,1~5:挑取的单菌落,0:阴性对照,5个单菌落均为阳性克隆,挑选其中两株活化保种;并挑选一株阳性转化子,提取质粒后命名为pcggαal-cas9,于-20℃保存。 二、重组质粒pcggαal-cas9转化产朊假丝酵母 1.阳性克隆的筛选 (1)将重组质粒pcggαal-cas9通过电穿孔转化法转入产朊假丝酵母并涂布含g418的ypd固体培养基,待菌落长出后将在含g418的ypd固体培养基上长出来的单菌落划线至一个新的含g418的ypd固体培养基上,30℃倒置培养过夜; (2)用无菌牙签挑选再次长起来的9个克隆分别接种于5ml含g418的ypd液体培养基中。培养48h左右提取酵母基因组,通过pcr扩增鉴定阳性克隆; (3)进行pcr反应,反应体系为100μl: 反应条件为: pcr反应结束后,通过琼脂糖凝胶电泳检测pcr扩增结果。电泳结果如图4所示,其中m:dnamakerdl2000,1~9:挑取的单菌落,0:阴性对照,表明9个单克隆均为阳性克隆,将重组菌株命名为cu-cas9. 2.rt-pcr检测cas9基因表达 挑选阳性重组产朊假丝酵母,接种于5ml含g418的ypd液体培养基中,培养48h左右,提取总rna。通过逆转录聚合酶链式反应(rt-pcr),以rna为模板经逆转录反应产生cdna,再以cdna为模板进行pcr扩增从而检测cas9基因是否表达。 (1)cdna第一条链合成: 首先从制备好的总rna中去除基因组dna。在无rnase的pcr管中加入以下试剂: 将pcr管置于37℃反应30min。然后加入1μl50mm的edta在65℃反应10min。将制备好的rna作为反转录的模板。 (2)第一链cdna合成 在pcr管中加入以下组分(先将rna模板与rnase-freewater混匀,65℃孵育5min后,冰浴2min,然后加入其他反应组分): 轻轻混匀各组分,在25℃孵育10min后,于42℃再孵育30min;85℃加热5s终止反应。按照以下体系继续进行pcr反应: 分装2管,反应条件为: 反应结束后,电泳检测pcr结果,结果如图5所示,其中m:dnamakerdl2000,1:pcr结果,电泳结果表明cas9基因在产朊假丝酵母细胞内可正常转录表达。 3.pcggαal-cas9质粒拷贝数鉴定 本实验采用相对定量法,以产朊假丝酵母木糖醇脱氢酶基因(xylitoldehydrogenase,xdhl)作为对照来测定cas9基因在产朊假丝酵母中的的拷贝数。通过比较cas9基因与产朊假丝酵母基因组上xdh1基因的相对含量关系,对cas9拷贝数进行相对定量。挑选阳性重组产朊假丝酵母,接种于5ml含g418的ypd液体培养基中。培养48h左右,提取基因组dna。参照荧光定量pcr试剂盒的说明书,按照下表配置pcr体系: 反应条件为: 所述xdh1基因的引物如表2所示: 表2 考虑到非理想状态下引物的扩增效率有可能达不到理论值,即100%,故将基因组按照10倍梯度进行8次稀释之后,分别作为模板进行扩增,并测定引物的实际扩增效率,计算cas9基因的相对拷贝数,荧光定量pcr结果如表3所示,表3表示了重组酵母cu-cas9中cas9基因的拷贝数,在重组产朊假丝酵母cu-cas9中,cas9基因的拷贝数是基因组的8.22倍。 表3 三、构建sgrna表达质粒 1.质粒的提取 将分别带有p426-crrna和pcuars-gapzαa的大肠杆菌接种到含有氨苄青霉素和博来霉素的lb培养基中,37℃,200rpm培养过夜,提取质粒。 2.产朊假丝酵母trnaglu基因的扩增 根据ncbi上公布的产朊假丝酵母基因组序列,设计特异性引物扩增产朊假丝酵母编码谷氨酸(ctc密码子)的转运rna,trnaglu基因,在下游引物cu-glu-r-srna的5’端加上bsmbi序列(斜体),以作为重叠pcr的引物,序列如下: cu-glu-f-psti: ctggagctgaattctgcagaatctcacaacaacaagaaaaatccggc cu-glu-r-srna: 使用takara高保真dna聚合酶扩增,反应体系如下: pcr反应条件 在以产朊假丝酵母基因组为模板扩增得到trnaglu后,进行琼脂糖凝胶电泳,如图6所示,其中m:dnamakerdl2000,1~2:pcr结果,扩增片段的大小约为100bp,与预期长度一致。 3.高保真酶扩增sgrna下游表达单元 通过引物bsmbi-f和cr-rna-r以p426-crrna为模板扩增得到sgrna下游表达单元(包括sgrna的结构区以及转录终止子区)。引物序列如下: bsmbi-f:ggaacgagacgctcgtctcggttttagagctagaaatagcaag cr-rna-r:gttatggatccggtaccttgactagtggccgcaaattaaagccttcg 使用takara高保真dna聚合酶扩增,反应体系如下: pcr反应条件 上述体系分别扩增两个目的片段后,进行琼脂糖凝胶电泳,再进行pcr产物胶回收。 4.重叠pcr 将上述得到的两个片段以重叠pcr连接起来,引入bsmbi酶切位点。反应体系如下: pcr反应条件如下 按照上述反应程序进10个循环之后,分别加入glu-f-psti和cr-rna-r各1μl,按照上述程序继续运行30个循环。待反应结束之后,取pcr反应液5μl,进行琼脂糖凝胶电泳,查看pcr反应结果。之后,纯化pcr产物,-20℃保存。 5.sgrna表达质粒的构建: (1)提取质粒pcuars-gapzαam,然后用限制性内切酶psti和bamhi酶切pcuars-gapzαam质粒和上一步骤中的重叠pcr产物,反应体系为50μl: 37℃水浴1h后,琼脂糖凝胶电泳分析酶切结果并保存,胶回收sgrna表达盒片段和pcuars-gapzαam骨架。 (2)sgrna表达盒片段与质粒pcuars-gapzαam骨架的连接 将回收到的sgrna表达盒片段和pcuars-gapzαam骨架按照摩尔比10∶1的比例进行连接,连接体系为20μl: 22℃连接1h后,连接产物转化大肠杆菌,涂布在含博来霉素的lb平板。 (3)阳性转化子的鉴定 待平板上长出转化子后,用无菌的牙签挑取少量菌体混于预先配置好的20μlpcr反应体系中,以重叠pcr的两条外引物鉴定阳性克隆。其中,阴性对照不含模板。反应体系为: 然后分装,每管20μl。反应条件为: 鉴定结果如图7所示,其中m:dnamakerdl2000,1~7:挑取的单菌落,从图中可以看出,挑选的7个克隆中,1-7均为阳性克隆,保存阳性克隆,并挑选阳性克隆经测序验证序列正确性,载体命名为pglu-sgrna。 四、识别产朊假丝酵母pgk启动子sgrna表达载体的构建 1.根据pgk启动子的序列,如序列表所示,通过程序筛选,选定pgk启动子序列(648-667)20bp作为靶点,序列为:agcaggcctacaattacggatgg(下划线为pam)。靶点序列及互补序列的5’端分别加上bsmbi酶切产生的粘性末端(小写字母),引物序列如下: 靶点序列:5’-ggaaagcaggcctacaattacgga-3’ 互补序列:5’-aaactccgtaattgtaggcctgct-3’ 2.以bsmbi酶切pglu-sgrna质粒,然后进行琼脂糖凝胶电泳以及胶回收,得到不含靶位点的pglu-sgrna骨架 3.将pgk的靶点序列和互补序列通过退火形成带有5’突出粘性末端的双链片段,将其与骨架连接,将连接产物转化大肠杆菌dh5α,待含博来霉素的lb平板长出单菌落,随机挑选单菌落进行pcr鉴定;如图8所示,其中m:dnamakerdl2000,1~6:挑取的单菌落,0:阴性对照,1-6均为阳性克隆,所得载体命名为pglu-sgrna-pgk。 五、gfp同源重组供体片段的构建 实验室前期构建的gfp表达载体带有产朊假丝酵母pgk启动子、gfp编码区和aoxtt终止子。为了将gfp基因通过同源重组整合到产朊假丝酵母的基因组的pgk位点,需要先将原gfp表达盒的pgk启动子5’端的一部分连接到aoxtt终止子3’端,从而作为重组片段的下游同源臂,如图9所示,其中a为改造前gfp表达盒结构示意图,b为供体片段pgk-gfp的结构示意图: 1.引物设计 利用软件primerpremier5.0设计扩增设计扩增pgk启动子5’片段的引物(f2、r2)和gfp表达盒其余部分的引物(f1、r1)。其中,f2的5’端加上r1的反向互补序列,以作为重叠pcr的引物。各引物序列如下: f1:ggtaccatccgtaattgtaggcctgc r1:gcacaaacgaaggtctcacttaatcttct f2: agaagatt4agtgagaccttcgtttgtgcttgtcttttaggagcttcttttcaccctg r2:ggtaccattgcagaatcattttacaatcgct 2.含有pgk左右同源臂dna片段gfp表达盒的扩增 以pcuars-pgk-gfp质粒为模板,使用特异性引物f1和r1扩增pgk启动子的3’片段至aoxtt终止子区域;f2和r2扩增pgk启动子5’端的648bp片段。pcr产物经琼脂糖凝胶电泳检测,然后胶回收纯化2个pcr产物;以f1和r2为引物,将此两个pcr产物通过重叠pcr连接到一起,形成完整的gfp同源重组片段。重叠pcr产物经加‘a’反应后克隆至pmd-19t载体上,转化大肠杆菌dh5α,涂布在含氨苄青霉素的lb平板上;待长出单菌落后用进行pcr鉴定,结果如图10所示,其中m:dnamakerdl8000,1~7:挑取的单菌落,可以得出1-7均是阳性克隆;挑选其中阳性克隆进行测序鉴定序列准确性,经测序验证的质粒命名为pmd19t-pgk-gfp。 3.pcr扩增同源重组供体片段 以pmd19t-pgk-gfp质粒为模板,使用f1和r2引物扩增gfp同源重组供体片段,扩增后进行琼脂糖凝胶电泳、纯化pcr产物,-20℃保存。本实施例中以pmd19t-pgk-gfp质粒为模板,使用f1和r2引物扩增含有pgk同源臂的gfp同源重组片段,大小为2488bp,琼脂糖凝胶电泳后结果如图11所示,其中m:dnamakerdl8000,1~2:pcr结果,扩增片段经纯化后用于转化产朊假丝酵母。 六、sgrna质粒和供体片段转化产朊假丝酵母 将pglu-sgrna-pgk质粒和同源重组供体片段pgk-gfp分别共转化野生型产朊假丝酵母和携带cas9表达载体的产朊假丝酵母,将转化后的野生型产朊假丝酵母和携带cas9载体的产朊假丝酵母分别涂布在含有博来霉素以及含有博来霉素和g418的平板上,30℃恒温培养箱倒置培养3-5天。挑取长出的单菌落,提取产朊假丝酵母的基因组鉴定gfp的同源重组情况。本实施例中在转化产朊假丝酵母后,各随机挑选了10个单菌落,提取基因组利用整合位点和pgk-gfp片段序列进行pcr鉴定,结果如图12所示,其中a为野生型产朊假丝酵母;b为表达了cas9的产朊假丝酵母,m为dnamakerdl8000,1~9为挑取的单菌落,0为阴性对照,可以得出野生型酵母的9个克隆中只有1个阳性克隆,为3号菌株;而携带cas9的酵母的9个克隆中8个为阳性,阳性率大大高于野生型。挑选三个阳性转化子进行测序验证,验证结果与预期一致,gfp正确整合到了预期的pgk启动子位点。结果提示cas9在产朊假丝酵母中发挥了作用,介导了gfp的同源重组,阳性重组子命名为cu-cas9-pgk-gfp。 七、gfp绿色荧光蛋白高表达菌株的筛选 1.荧光显微镜的检测 随机挑选宿主为表达cas9蛋白的产朊假丝酵母的gfp阳性转化子(cu-cas9-pgk-gfp)接种于含有博来霉素和g418的ypd液体培养基中,30℃,200rpm培养24h。取适量菌液适当稀释后,置于荧光显微镜下分别进行观察,以转化前的产朊假丝酵母作为对照。结果如图13所示,其中a为明场下cu-cas9,b为蓝色激发光下cu-cas9,c为明场下cu-cas9-pgk-gfp,d为蓝色激发光下cu-cas9-pgk-gfp;在蓝色光激发下,产朊假丝酵母原始菌不发荧光,而整合了pgk-gfp基因的的产朊假丝酵母细胞发出绿色荧光,表明gfp在产朊假丝酵母中正常表达。 2.流式细胞仪检测 (1)挑选宿主为表达cas9蛋白的产朊假丝酵母的gfp阳性转化子接种于含博来霉素和g418的ypd液体培养基中,30℃,200rpm培养60h; (2)每隔12h取适量菌液于4℃,10,000×g离心60s后,用1×pbs液重悬菌体,使菌液的od600在0.1-0.3; (3)以转化前的产朊假丝酵母作为对照组,用流式细胞仪bdaccuritmc6检测gfp的平均荧光强度。 本实施例中,挑选三株阳性转化子转接至含博来霉素和g418的ypd液体培养基中,30℃,200rpm培养60h,以产朊假丝酵母原始菌作为对照,用流式细胞仪分别检测各阳性转化子的平均荧光强度。结果如图14所示,其中gfp-0为原始菌cu-cas9,gfp-1、gfp-2、gfp-3均为阳性重组菌cu-cas9-pgk-gfp,重组产朊假丝酵母的平均荧光强度在培养48h左右达到最大值,远高于对照组的平均荧光强度。这进一步验证crispr-cas9系统成功介导gfp基因整合到了产朊假丝酵母基因组,并且gfp基因表达良好,说明crispr-cas9系统应用于产朊假丝酵母外源基因的定点插入是可行的。 八、菌株筛选标记的丢除 crispr-cas9系统的一个优势是可以实现无选择标记导入外源基因。为实现这一目的,在挑选出成功表达gfp的工程菌之后,通过将工程菌在无抗生素培养基中培养,使其丢失游离质粒,实现抗性基因的去除。与酿酒酵母相似,产朊假丝酵母增值一代所需的时间约为1.5h,经过96h后产朊假丝酵母将近传64代 (1)挑选高表达绿色荧光蛋白的菌株,将其接种于不含抗生素的ypd培养基中,30℃,200rpm,培养过夜。 (2)将活化后的菌株以2%接种于不含抗生素的ypd培养基中,30℃,200rpm,培养过夜。 (3)重复步骤2直至96h(传代64代左右),将菌株取适量菌液进行稀释后涂布于不含抗性的ypd培养基平板上,30℃倒置培养。 (4)待平板上菌落形成后,挑30个单菌落分别点种于无抗生素,含g418,含博来霉素的ypd培养基平板上,30℃倒置培养3天,结果如图15所示,其中a为含有博来霉素的培养基,b为含有g418的培养基,c为不含抗生素的培养基;从图中可以看出重组酵母在ypd平板上的生长情况,通过96h的传代培养,在无选择压力的情况下,游离质粒部分缺失,经过挑选可得到不含任何筛选标记的重组菌株。 (5)挑选只在无抗生素平板上生长,而在含g418和博来霉素的ypd培养基上不生长的菌落用无菌牙签接种于无抗生素的ypd液体培养基中,待菌落长起来后保种。 九、无抗性基因阳性菌株gfp表达水平的检测 随机挑选已去除抗性基因的阳性转化子接种于不含抗生素的ypd液体培养基中,30℃,200rpm培养60h,期间取样用流式细胞仪分别检测各克隆的平均荧光强度,结果如图16所示,其中gfp-0为原始菌cu-cas9,gfp-1、gfp-2、gfp-3为阳性重组菌cu-cas9-pgk-gfp,在去除抗性筛选标记后,重组菌株仍然能够正常表达gfp蛋白,而且gfp蛋白的表达水平与抗性丢除之前相差不大。进一步说明gfp基因整合到了酵母的基因组上,没有随着传代而丢失,可以稳定遗传到子代细胞。 实施例2 crispr-cas9介导果胶甲酯酶基因靶向插入产朊假丝酵母基因组 本实施例所用的表达载体与上述实施例1中的表达载体来源相同,引物合成及dna测序由invitrogen公司完成,产朊假丝酵母游离型表达载体:pcuars-pgkg-gfp,其具体结构及特殊序列如图26所示;带有pmea基因的质粒pcggαal-pmea(含有产朊假丝酵母的gap启动子,g418抗性基因),其具体结构和特殊序列如图27所示。pglu-sgrna-pgk质粒及含有cas9的产朊假丝酵母均为实施例1中构建完成的产物;引物序列-f代表上游引物,引物序列-r代表下游引物。 基于已有的pglu-sgrna-pgk质粒和含cas9的产朊假丝酵母,需进行含左右同源臂的重组供体片段构建。 一、pmea同源重组供体片段的构建 1.将pmea游离型表达载体的gap启动子换成pgk启动子 分别提取pcggαal-pmea和pcuars-pgkg-gfp质粒,用bsp119i和bglii分别对两个质粒进行双酶切后,将pgk启动子与载体pcggαal-pmea进行连接,将产朊假丝酵母pgk启动子片段替换pcggαal-pmea载体的gap启动子后,转化大肠杆菌dh5α,涂布在含有卡那霉素的lb平板上过夜培养;待长出单菌落后,挑选6个菌落,以pgk启动子5’端片段引物(f2和r2)进行菌落pcr鉴定,鉴定结果如图17所示,其中m:dnamakerdl2000,1~6:挑取的单菌落,结果显示pgk启动子已连接到pmea载体上,将其命名为pcpgαal-pmea。 2.含有pgk同源臂的pmea同源重组片段的构建 参照实施例1中构建gfp同源重组片段的方法,以pcpgaal-pmea质粒为模板,使用特异性引物f1和r1、f2和r2分别扩增pgk启动子3’片段至aoxtt3’末端长度为1800bp的dna片段以及pgk启动子5’端长度为648bp的dna片段。两个pcr产物经纯化后以重叠pcr将它们连接起来,形成完整的pmea同源重组片段,同源重组片段的结构如图18所示。pmea同源重组片段经加‘a’反应后克隆至pmd19-t载体,测序验证序列正确性,本实施例中将重组片段连接pmd19-t载体后涂布在含氨苄青霉素的lb平板上,待平板长出单菌落后用m13通用引物进行pcr鉴定,结果如图19所示,其中m:dnamakerdl8000,1~7:挑取的单菌落,0:阴性对照,从图中可以看出除阴性对照外,其余克隆和阳性对照扩增出单一且清晰的目的条带,表明供体基因片段pgk-pmea已成功克隆入t载中;挑取三株阳性转化子活化后,取1ml菌液送至invitrogen公司进行测序,并将测序结果与已知的基因序列进行对比,结果显示阳性克隆中的pgk-pmea片段核酸序列与已知序列相似性为100%,将经测序验证正确的菌株命名为pmd19t-pgk-pmea。 二、sgrna质粒和供体片段转化产朊假丝酵母 以pmd19t-pgk-pmea质粒为模板,使用f1和r2引物扩增pmea同源重组供体片段pgk-pmea;将pglu-sgrna-pgk质粒和重组同源片段pgk-pmea共同转化表达cas9的产朊假丝酵母cucas9;电转后,将菌体涂布于含有博来霉素和g418双抗生素平板上,30℃恒温培养箱倒置培养3-5天。挑取长出的单菌落进行活化,提取基因组后以pcr鉴定阳性克隆,结果如图20所示,其中m:dnamakerdl8000,1~9是九个克隆的pcr结果,0是阴性对照,可以看出,条带1、2、4、5、6、7、9表明为阳性克隆,将阳性克隆进行活化保种。 三、高果胶甲酯酶酶活菌株的筛选 1.挑选3株阳性重组菌株接种于5ml含有博来霉素和g418的ypd液体培养基中,30℃,200rpm条件下培养过夜后,转接至新的100ml含有g418和博来霉素的ypd液体培养基中,培养96h之后取4ml菌液,12,000×g离心4min,吸取上清液,用来检测果胶甲酯酶酶活,选择一株果胶甲酯酶酶活最高的酵母重组菌用于后续的实验。本实施例结果如表5所示,表5展示了三株重组菌上清液中果胶甲酯酶的酶活,可以看出pmea-3重组菌的上清中果胶甲酯酶酶活最高为7.9u/ml,因此将pmea-3重组菌用于后续实验研究。 2.果胶甲酯酶酶活的测定 果胶甲酯酶酶活的测定采用氢氧化钠滴定法,其原理是果胶甲酯酶在果胶脱酯过程中会产生甲醇和果胶酸,用标准naoh溶液滴定生成果胶酸的羧基,根据消耗的naoh的量来计算果胶甲酯酶的酶活。酶活测定方法具体步骤如下: 按表4进行配置,表4为测果胶甲酯酶酶活所需试剂,于50ml的锥形瓶中依次加入ph5.0的磷酸氢二钠-柠檬酸钠缓冲液、1%的果胶溶液、酶液,50℃反应40min后沸水浴5min终止反应。冷却至室温后,以酚酞为指示剂,用10mm的naoh标准液进行滴定。使用灭活的酶液作为空白对照,每组做3个平行。 果胶甲酯酶酶活力单位的定义为:在50℃,ph5.0的条件下,每1min释放1μmol羧酸所需要的酶量为1个酶活力单位,用u/ml表示。 酶活力的计算公式可以表示为: 式中c:果胶甲酯酶的酶活,u/ml;n:稀释倍数; v1:消耗的氢氧化钠体积,ml;v2:酶液体积,ml; 10:naoh标准液的浓度,mm;t:反应时间,min。 表4 3.果胶甲酯酶的pmea的sds-page分析 将高果胶甲酯酶酶活菌株的筛选步骤1中30℃、200rpm条件下培养96h的菌液离心收集上清液后,按上清液∶无水乙醇体积比1∶3的比例于上清液中加入无水乙醇,-20℃处理30min沉淀蛋白质,然后12,000×g离心10min收集沉淀用于sds-page分析。 (1)sds-page凝胶配制,配置tris-甘氨酸sds-page所用溶液如表5所示: 表5 (2)样品处理: 按以下体系配制电泳样品,沸水煮5min后,8,000×g离心3min; 蛋白样品:800μg 5×loadingbuffer:8μl ddh2o:补足至40μl (3)点样:处理后的蛋白样品上样量12μl,蛋白marker上样量为5μl; (4)电泳:电压设置为100v,电泳2h; (5)染色及脱色:电泳后将凝胶从胶板上取下来,放入考马斯亮蓝染色液中染色1h;沸水浴脱色约20min,至条带变清晰。 结果如图21所示,其中m:10~170kda的蛋白marker,1:pmea-3胞外上清液,2:cu-cas9胞外上清液,考马斯亮蓝染色结果显示在40kda~55kda之间出现了条带,高于果胶甲酯酶蛋白预期的分子量37kda,这可能是由于重组的果胶甲酯酶发生了不同程度的糖基化修饰造成的。 四、重组菌的生长曲线和酶活曲线 将高果胶甲酯酶酶活的产朊假丝酵母重组菌参照实施例1的方法丢弃抗性选择标记基因后,活化接种于5ml不添加任何抗生素的ypd培养基中,在30℃,200rpm,培养24h后,再按2%接种量转接至50ml新鲜的不含抗生素的ypd培养基中,培养96h。每隔12h取样4ml分别测定菌体密度(od600)及上清液中的果胶甲酯酶的酶活。以时间为横坐标,分别以od600和酶活(u/ml)为纵坐标绘制重组菌的生长曲线及酶活曲线。本实施例中将上述pmea-3菌株在无抗生素的ypd液体培养基中传代培养96小时后,涂布于不含抗生素的ypd平板上生长。待平板上菌落形成后,挑50个单菌落分别点种于不含抗生素、含有g418抗生素、含有博来霉素抗生素的ypd平板上培养3天,结果如图22所示,其中a:含有博来霉素的培养基;b:含有g418的培养基;c:不含抗生素的培养基,从图中可以看出,通过96h的传代培养,在无选择压力的情况下,游离质粒会丢失,经过筛选得到不含任何筛选标记的能正常表达果胶甲酯酶的重组菌株,命名为cu-pgk-pmea。 获得无抗性筛选标记的重组菌株后,通过测定重组产朊假丝酵母菌cu-pgk-pmea的生长曲线和酶活曲线来评估pmea的表达情况,结果如图23所示。从图中可以看出,重组产朊假丝酵母菌cu-pgk-pmea在0h~6h处于迟缓期,6h~36h处于对数生长期,36h~48h是稳定生长期,48h时od600达到最大值30.1,48h之后菌体处于衰亡期。果胶甲酯酶酶活曲线与生长曲线相对应,在12h~48h期间酶活处于增长状态,60h时达到最大值为8.3u/ml,60h之后酶活有所下降。菌株的生长曲线和酶活曲线表示,外源基因的表达对菌体的生长状态影响不大,表达果胶甲酯酶重组菌株od600的最高值是30.1。果胶甲酯酶的最高酶活为8.3u/ml,是现有技术中毕赤酵母中表达果胶甲酯酶最高酶活1.1u/ml的8倍左右。通过在无抗生素的ypd培养基中传代培养96h,成功的去除了重组菌株的pcggαal-cas9和sgrna质粒,得到了不含抗性基因、高效表达果胶甲酯酶的产朊假丝酵母,进一步验证了利用cas9系统能够介导目标基因插入至产朊假丝酵母中,并提供了构建一种产朊假丝酵母表达系统的思路。 显然,本发明的上述实施例仅仅是为清楚地说明本发明技术方案所作的举例,而并非是对本发明的具体实施方式的限定。凡在本发明权利要求书的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。 序列表 广东利世康低碳科技有限公司 广州利世康工业科技有限公司 暨南大学 一种验证crispr/cas9系统介导目标基因插入产朊假丝酵母的可行性的方法 2 siposequencelisting1.0 1 23 dna 未知(unknown) 1 agcaggcctacaattacggatgg23 2 1340 dna 未知(unknown) 2 ttgtcttttaggagccttcttttcaccctggctttcttcagactccacgcctctcgcccg60 tttgttgttgatcttcttctgttgcttcttcgtgagcttaccagtatccagatgcgttgt120 cagggcaagagggtcatgttcaagctcctctttcactttcagtccaatacgtttccaggc180 agggatgtgttcgctcatcgttccagactcgagtggtgaaaactatggcaacctctactt240 cctttccaaacacacagcgtgctttgtagtgtgtgcctaagagctgaattttttttcctt300 ccatgctgcgctgcgatgagctctgcccgcccgcagcctcggaggctagcgacgtataaa360 aaaggcctgtgaaaattttatcctcctccttaacgacccttctttctcttcttcacattc420 aaaaacttcaagcagctgtctctgttcctttgctgtgttctaccattggatattcccatt480 ccccgtggagaaccgaactggagtctagcagcatgcgagatcaatattacacggtttgag540 tctgatacgcttgagcagccattttttggcttctcctggtgtgtatccagatatagaagt600 tcgtatacatttcccatagcgattgtaaaatgattctgcaatggaaccatccgtaattgt660 aggcctgctgagatggcactcgcaatgcctctgtgtctggttttttgccttctccgtcca720 tcagcaccagtggcttcttagggcataacgagacggctccttggtgaaagatgccctgct780 ccgtctgtctgcctgttgctacaaccactgcgtagtcagatgacccggtctgtgtgctgt840 ggaatcaccgggagcgaaattccggtttcgctggcagatgagctcatcaaccacatcaac900 tggagcaacctcaccagaggacacgtaacctgcccggttgaattctgtcaaaccgtacat960 cacacaacaacagcagcagcaacaacaacaacgtcagttgtcgttcgcatggcgacgtta1020 cctaacggcaccaacatcgtctcgtcctcgccagtgcctgtttcccctacccggagtggc1080 ccggcccacctgtcgttcttttttcgtcaattgtgtccagctggtgccatcaccatatgt1140 tcaagtgcgtggcctgtactagcgcagtctgctgcagtataaaagggattgctgaggccc1200 cctttagcgtttccaattaacaattgattcccttttccccatagtccgtttgtactacat1260 cctacataacaaaagtgagtgttacaagacaagtgtggcggtcaattggatcatttggac1320 taacatactggcggataaag1340 |
【本文地址】