CSV合规的矫枉过正 |
您所在的位置:网站首页 › csv验证 › CSV合规的矫枉过正 |
CSV合规的矫枉过正
|
导读 曾几何时,当KillerParker用一封又一封警告性教育了中国和印度医药企业CSV的重要性之后,药企突然发现自己要给全世界科普起什么是GMP,想想你有没有遇到过这样的场景 探讨非GMP第三方、IT开发部门直接参与GMP业务的可行性方法背景和结论 1.请软件公司为自己配置权限–100多个权限,6个用户等级;工程师按照用户需求勾选好了权限,但是验证工程师却要求将所有权限都测试一次,甚至是正向反向,外加挑战性测试。别的行业一本标准实施方案报告,药企就能整出十本的厚度;理由还是基于风险,必需挑战系统功能的可靠性? 2.考虑集团公司GMP管理更合规高效,将培训搬到线上,将数据云存储–但是面临检查的时候,检查官却挑战数据存在公司之外,不满足数据可靠性标准,GMP法规也未规定允许这样做;可是GMP法规确实未能明文规定,但是也并没有明文禁止;而且考虑到数据安全与业务连续性,不可否认GMP数据存在阿里巴巴云端服务器的安全性远远高过一般药企的简陋IT机房,哪一个数据可靠性风险大? 总之这篇文章探讨的一个话题–GMP环境中,CSV-ComputerizedSystemValidation计算机化系统的合规不应该被特殊化,如果我们引入其他行业的先进技术(如IT信息化,工程自动化)来提升医药GMP生产和管理水平,不应该一味要求药企按照GMP验证套路,重新论证一次对方的技术和系统,而应该思考对方行业中原有的管理体系中是否有和质量管理一脉相承的体系可以直接引用(如软件开发生命周期管理,ISO27001等)。重复工作意味着额外无效成本,这显然和国家4+7政策导向想违背的;更遑论既然非专业的药企,真的可以通过学习(套用)GMPCSV,去检验专业的软件公司,IT企业开发的系统的可靠性吗? 化工厂专业生产化工品,药企主导审计来评估其是否适合提供医药原料;IT公司专业提供计算机化系统(如ERP、自动仓等)和服务(云存储,灾难恢复等),药企也应该可以依靠审计来判断是否满足自身GMP业务需求。 正文 随着“自动化和信息化-两化融合”,“工业4.0智能制造”和“CSV”等这些趋势在制药行业的兴起,越来越多的GxP信息化系统(如ERP,LIMS,MES,EDMS)以及自动化系统(如SCADA,PCS,DCS)等电子管理系统开始在GMP合规上担当越来越重要的角色(如图一)。
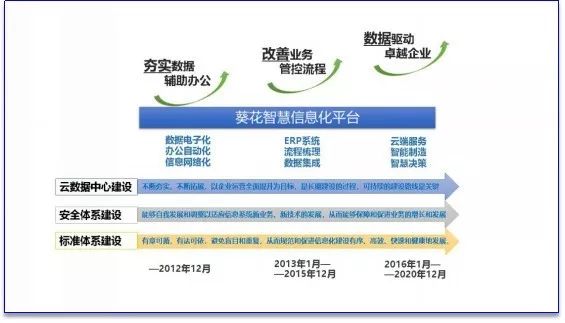
图一。中国葵花药业数字化转型,逐渐向GMP生产延伸 与此同时,作为推动这一行业趋势变化的主力之一,IT部门或外部第三方技术公司,越来越多地参与到药品GMP生产过程中同时(如图二),也越发受到监管部门重视,甚至像之前API供应商一样要求制药企业将这些技术部门纳入GMP管理范畴以满足GMP合规要求。而事与愿违,某些时候培训IT或者第三方技术服务商GMP知识,使之合规–非常不易!
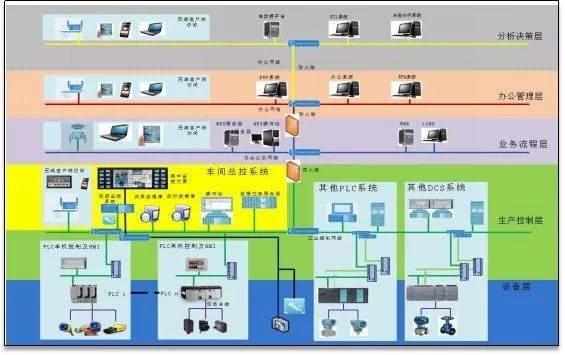
图二。从制造生产到实验室检验,计算机化系统无处不在 本文也将就“GxP系统服务供应商管理”这个需求进行讨论,分析如何跨越行业差异(自由多变的IT技术行业与严格监管的医药行业),探讨非GMP技术部门参与GMP业务的可行性方法。
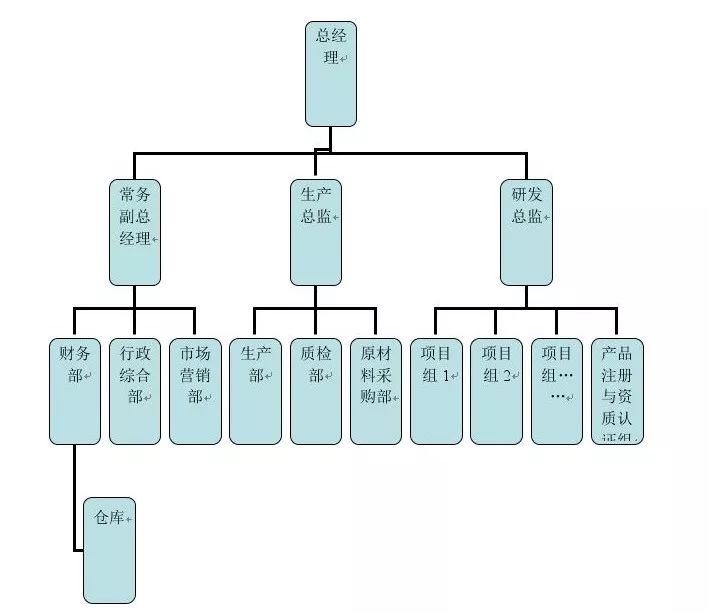
图三。传统GMP药厂组织架构中,IT信息部一般直属于总部,不属于GMP部门 国外法规要求 参考 EU GMP 附录 11 计算机化系统 –Chapter 3 Supplier and Service Provider 法规原文关键词3.1 When third parties (e.g. suppliers, service providers) are used e.g. to provide, install, configure, integrate, validate, maintain (e.g. via remote access), modify or retaina computerised system or related service or for data processing, formal agreements must exist between the manufacturer and any third parties, and these agreements should include clear statements of the responsibilities of the third party. IT-departments should be considered analogous.3.1当第三方(如供应商、服务提供商)使用如提供、安装、配置、集成、验证、维护(例如通过远程访问),修改或保留一个计算机化的系统或相关服务或数据处理,厂商之间的正式协议必须存在和任何第三方,和这些协议应包括明确的说明责任的第三方。公司内部IT部门也不例外。formal agreements – 正式的合约,一般是 Service Level Agreement 服务水平协议; 定义第三方的服务内容及标准(如提供、安装、配置、集成、验证、维护);注意同一个公司的 IT部门也适用这个条例;3.2 The competence and reliability of a supplierare key factors when selecting a product or service provider. The need for an audit should be based on a risk assessment. 3.2供应商的能力和可靠性是选择产品或服务供应商的关键因素。审计的需要应以风险评估为基础。an audit – 必要的审计检查,即通过正式的审查来判断评估技术部门的能力和可靠性是否满足 GMP业务需求;3.3 Documentation supplied with commercial off-the-shelf products should be reviewed by regulated users to check that user requirements are fulfilled. 3.3 相应GMP用户应审查随商业现货产品提供的证明文件,以检查用户要求是否得到满足。Reviewed ….to check that user requirements are fulfilled. – 回顾审核供应商或者服务商随计算机化系统提供的技术文件(用户手册,安装确认方案报告),来确认系统是否满足需求;3.4 Quality system and audit information relating to suppliers or developers of software and implemented systems should be made available to inspectors on request.供应商或者软件系统开发商的 QMS和审计信息,应该可以作为证据提供给检查官Quality system and audit information …available to inspectors on request – 如果 GMP检查有需要,需要提供服务提供商相关资料(质量系统 Quality System和审计报告)
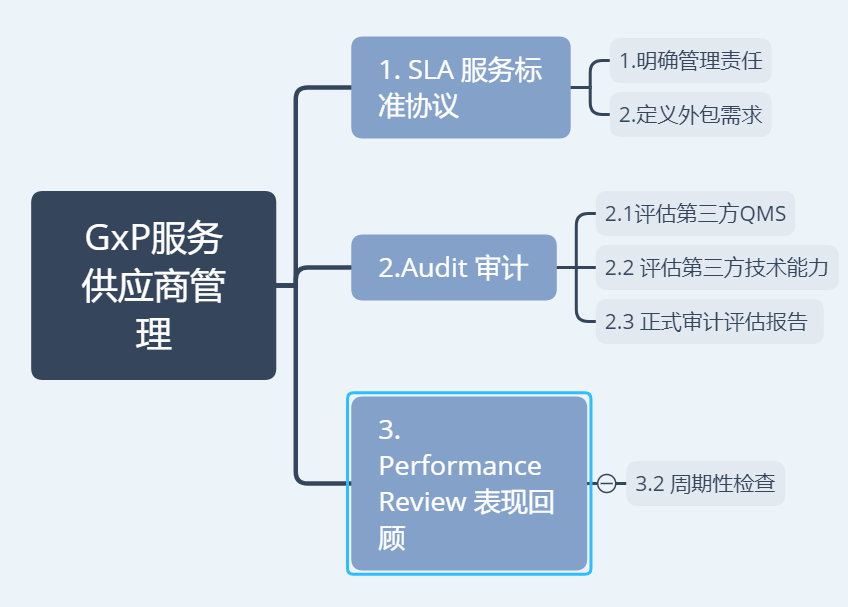
实施路径
如上图所示: 1.1明确管理责任:正如ICHQ10中对外包服务的监管要求规定–“Evenwheretasksarepartlycontractedouttoexternalcompanies,theregulateduserisultimatelyresponsibleforensuringthesuitabilityandoperabilityofthecomputersystems.”如果制药公司某些GMP部门将其GxP系统的设计、验证和运维外包给第三方(或者集团总部IT部门),对于GxP系统的合规性和可用性的最终责任还是归属GMP用户部门本身,而这些部门也必需有义务,同时有方法去保障GxP系统和其数据的可靠性。 1.2定义外包需求:制药公司可以通过签署正式合约或者内部备忘录的方式来管理第三方服务商的业务支持行为,双方应该在这份文件中明确各自的责任义务的同时,也应定义清楚外包需求(如系统运维,数据备份,用户管理,灾难恢复,审计支持等),确保对各自工作范围清晰无异议。 2.1审计评估第三方QMS:无论是实体设备仪器,还是无纸化信息系统,其良好质量保障都少不了背后相关的开发、测试、上线以及运维文件的支持。如同PIC/S要求,”CompliancewitharecognizedQualityManagementSystem(QMS)providestheregulateduserandregulatoryagenciesorcompetentauthoritywiththedesiredconfidenceinthestructuralintegrity,operationalreliability,andongoingsupportforsystem,softwareproduct,orsoftwareserviceutilizedinthesystem.[ii]”没有一个运行良好的QMS的背书,监管方很难认可其软件稳定性或者服务质量。 2.2审计评估第三方技术能力:基于ISO12207建议[iii],在正式外包重要系统的某些日常业务之前,受监管公司应从以下这些方面评估重要GxP系统某些业务外包给第三方这一行为是否适应: l外包系统基于病人安全和数据完整性潜在影响 l供应商的质量管理体系 l外包服务运行可靠性 l系统的新奇性或复杂性 l软件工程实践(SoftwareEngineeringPractice) l第三方提供类似服务的历史和经验 l对软件和硬件产品的持续支持能力 2.3正式审计评估报告–结合上述要求,审计过程发现和对应缺陷项的后续追踪应形成正式文件,签字批准留档供将来官方检查(EUGMPAnnex11-3.4)。正式的GxP服务供应商审计报告至少应包括以下内容: l确定受监管公司的目前GxP系统管理水平 l确定由于外包某些GxP系统业务员而给项目带来的潜在风险 l确定第三方外包服务商的质量管理体系的缺陷 l确定是否使用第三方的文件,以支持GxP系统验证与运维的合规性* l建立共同的质量理解/伙伴关系 *如果确认需要直接引用第三方文件,来满足GMP合规要求(如备份SOP,灾备方案的技术资料),QA和相关业务流程所有者需审核并签批第三方文件(ASTME2500-13)。 3.1周期性检查PerformanceReview–如果由第三方(外部供应商或内部IT)为GxP系统的某些业务提供服务(如系统运维,数据备份,用户管理,灾难恢复等),那么制药公司应该定期地回顾其提供服务的质量与及时性,以确保不会对患者安全,产品质量和数据可靠性产生不可挽回的影响。周期性回顾的频率可以通过风险评估,基于系统的重要性来进行评估。 综上所述,对于GxP系统的服务供应商合规性的管理,除了可以考虑将培训IT人员,将IT部门纳入GMP领域(如用户管理,偏差异常处理等);也可以因地制宜,通过SLA,Audit和PerformanceReview的方式,指引非GMP部门的第三方(如运维ERP和HR软件的总部IT,云服务供应商等)为GMP业务提供技术支持的同时,可以满足GxP系统的强监管要求,经得起官方检查的挑战! 以上见解属于笔者(蒲公英论坛-CSV胡大侠)个人见解,如有任何建议或者意见,欢迎来信邮件[email protected]讨论. 参考文献 ICHQ10,PharmaceuticalQualitySystems,June2008,Section2.7,ManagementofOutsourcedActivitiesandPurchasedMaterials. [ii]PI011-3.“GoodPracticesforComputerisedSystemsinRegulated‘GXP’Environments,”PharmaceuticalInspectionCooperationScheme(PIC/S),September2007. [iii]Evaluation—asystematicdeterminationoftheextenttowhichanentitymeetsitsspecifiedcriteria(ISO12207:1995).(Note:The1995revisionisnotthemostrecentversion.) |
【本文地址】
今日新闻 |
推荐新闻 |