【RNA |
您所在的位置:网站首页 › cpm格式转化 › 【RNA |
【RNA
|
在RNA-seq上游的流程中,所得到的产物为表达矩阵,一般指通过RSEM、HTseq等量化工具统计得到的,各个样本比对到参考基因组中各个基因的reads数,一般成为raw read count,这也是最简单的表达定量形式。 但是在分析不同样本中基因的表达量时,使用raw read count是不够严谨的,往往还需要进行表达矩阵的归一化处理,这种处理需要的解决的问题如下: 1.目标基因的转录本长度,长度越长的转录本对应能够mapping上的reads数也越多,无法反映该基因实际的表达情况 2.总的有效比对的reads数量,即去除没有mapping上的,或者匹配不准确的reads,显然测序深度越深,总的有效比对的reads数量越多,每一个基因对应mapping到的reads数量也越多 Coverage ratio(覆盖比率,亦简称覆盖率,亦称基因组覆盖率),指被测序到的碱基占全基因组大小的比率。 Coverage depth (覆盖深度,亦称测序深度,或者碱基平均测序深度),指每个碱基被测序的平均次数。即测序的数据总量比基因组大小 3.测序的偏差 方法一:RPKM RPKM: Reads Per Kilobase of exon model per Million mapped reads (每千个碱基的转录每百万映射读取的reads)
将raw counts除以改样本总的有效的(匹配上的)reads数(以KB为单位),以减少测序深度的影响;再除以该基因的转录本长度(以百万为单位),即真核生物外显子长度和、原核生物编码区长度和,以减少基因长度的影响 。 在single-end测序中,一个read就是一个read。而在pair-end测序中,若一对paired-read 都比对上了,当做两个read;若只有一个read比对上,另一个未比对上,当做一个read计算。 局限性:该算法除以有效比对的reads总和的这种方式,排除了测序深度对总reads数的影响,但是没有考虑到基因转录本长度对reads总和的影响,可以说RPKM是先进行测序深度标准化,后进行基因长度标准化。为了排除转录本长度对reads总和的影响,应当首先进行基因长度标准化,载根据长度标准化后的reads总和进行测序深度的标准化,TPM算法应运而生。 方法二:TPM TPM:Transcripts Per Kilobase of exon model per Million mapped reads (每千个碱基的转录每百万映射读取的Transcripts) TPM是先根据基因长度矫正count值,即用count值除以基因长度(百万为单位)得到矫正后的reads数,将所有校正后的count值相加得到矫正后的总counts值,将校正后的count值除以校正后的总count值,得到TPM值。 方法三:FPKM FPKM: Fragments Per Kilobase of exon model per Million mapped fragments(每千个碱基的转录每百万映射读取的fragments) 一般是针对双端测序的方法 在single-end测序中,FPKM将read当做fragment计算,此时FPKM和RPKM是相同的。而在pair-end测序 中, 若一堆paired-read 都比对上了,当做一个fragment。 方法四:RPM/CPM RPM/CPM: Reads/Counts of exon model per Million mapped reads (每百万映射读取的reads)
RPM(CPM)是指将count数除以总有效匹配的count数,以排除测序深度的影响,常常用于长度差异不大的sRNA的分析
参考链接:对FPKM/RPKM以及TPM的理解 - wangchuang2017 - 博客园 TPM、RPKM与FPKM相互转换 - 简书 (jianshu.com) RPKM简介 | Public Library of Bioinformatics (plob.org) |
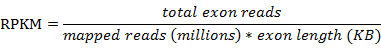

【本文地址】
今日新闻 |
推荐新闻 |