Antismash |
您所在的位置:网站首页 › big网络 › Antismash |
Antismash
|
文章目录
Antismash
概述
Install
使用
其他参数
输出详细结果
网络文件
GCF and GCC
交互式可视化
网页版
时间
其他详细参数
BigScape
简介
工作原理
install
usage
输出
输出文件夹结构
结果
可能的问题
Reference
Antismash
概述
antiSMASH - the antibiotics and Secondary Metabolite Analysis SHell,是用来鉴定微生物基因组次级代谢物合成基因簇的软件。 临床上使用的大部分抗生素和药物均来自植物或微生物的天然产物。结合基因组挖掘的经典分离与分析法使得基于基因组的天然产物途径鉴定和描述更为方便。 一般情况下,参与次级代谢途径中生物合成酶基因在基因组上成簇排列,基于指定类型的HMM,antiSMASH数据库能准确鉴定所有已知的次级代谢簇。在antiSMATH中,将次级代谢簇分为24类。 Install conda create -n antismash -c bioconda antismash conda activate antismash (antismash) ok@ubuntu $ download-antismash-databases Creating checksum of Pfam-A.hmm PFAM file present and ok for version 32.0 Resfams database present and checked Creating checksum of TIGRFam.hmm TIGRFam database present and checked Creating checksum of proteins.fasta ClusterBlast fasta file present and checked Creating checksum of proteins.fasta ClusterCompare mibig FASTA file present and checked Pre-building all databases... done. # antiSMASH 6.0.1 conda deactivate # 若出现 Could not remove or rename /home/anaconda3/pkgs/backports_abc-0.5-py_1*,手工删掉,尤其是.conda的文件,手工删掉 # sudo rm -rf /home/anaconda3/pkgs/backports_abc-0.5-py_1* # manually ## First add the antiSMASH debian repository: sudo apt-get update sudo apt-get install -y apt-transport-https sudo wget http://dl.secondarymetabolites.org/antismash-stretch.list -O /etc/apt/sources.list.d/antismash.list sudo wget -q -O- http://dl.secondarymetabolites.org/antismash.asc | sudo apt-key add - sudo apt-get update ## then install the binaries themselves sudo apt-get install hmmer2 hmmer diamond-aligner fasttree prodigal ncbi-blast+ muscle glimmerhmm 使用Fast run 在没有参数的情况下运行antismash将运行核心检测模块和所有快速的簇特定分析步骤。更多耗时的选项,如ClusterBlast分析、基于簇的PFAM注释、smCoG树生成等将不会被运行。在四核机器上,使用这些选项运行辅酶链霉菌基因组将需要大约两分钟。 从prokka注释好的gbk文件预测 (antismash) @ubuntu:/mnt/80G4/TGG_yut/580MAGs_antismash$ time antismash -c 10 --allow-long-headers --output-basename MGGQSC.20.08_metabat2_bin.2 ../580MAGs_prokka/MGGQSC.20.08_metabat2_bin.2.gbk &> MGGQSC.20.08_metabat2_bin.2.log & #18min 从组装好的基因组序列文件预测 (antismash) @ubuntu:/mnt/80G4/TGG_yut/580MAGs_antismash$ time antismash -c 10 --cc-mibig --genefinding-tool prodigal --output-basename MGGQSC.20.08_metabat2_bin.2_genome ../580Batch2_High_quality_MAGs/MGGQSC.20.08_metabat2_bin.2.fa &>MGGQSC.20.08_metabat2_bin.2_genome.log& # --genefinding-tool 必须要指定 # 20min # 即使是gbk文件也最好指定--genefinding-tool,因为某些record可能没有CDS区域 nohup time antismash --genefinding-tool prodigal -c 10 --cc-mibig --output-basename GLYI1.20.09_maxbin2_bin.14 /mnt/80G4/TGG_yut/580MAGs_prokka/GLYI1.20.09_maxbin2_bin.14.gbk & # 注意--output-dir目录必须是空的 统计长度 (base) [yutao@myosin antismash_links]$ for i in *gbk;do len=$( head -n1 $i| awk '{print $3}');printf $i"\t"$len"\n";done|sort -k2 -nr >../184OTUs_antismash_BGCs_length.tsv 结果:当前目录下生成basename的目录,所以记得在输出目录运行脚本 打开index.html,可以看到2个BGC区域,包括区域、BGC类型以及基因组的位置 打开index.html,可以看到2个BGC区域,包括区域、BGC类型以及基因组的位置  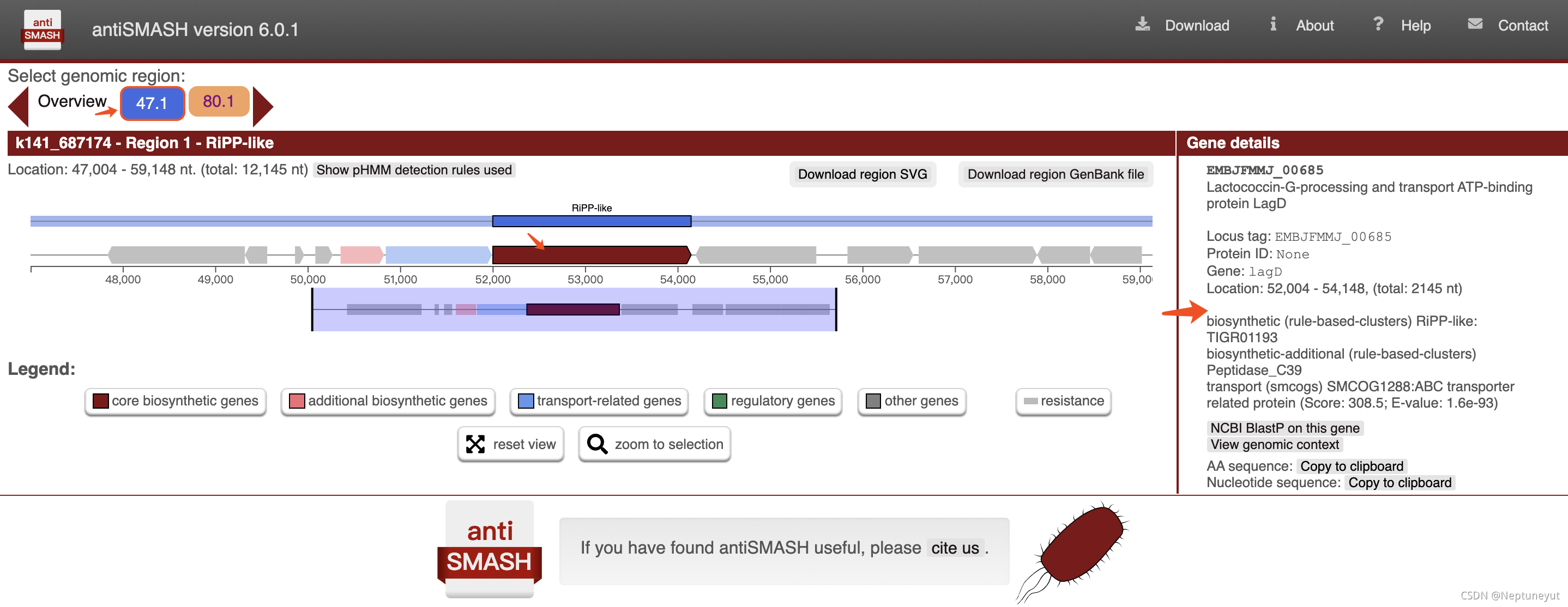 如果加上–cc-mibig,则会在最下面展示MiBIG数据库hit到的情况
如果加上–cc-mibig,则会在最下面展示MiBIG数据库hit到的情况 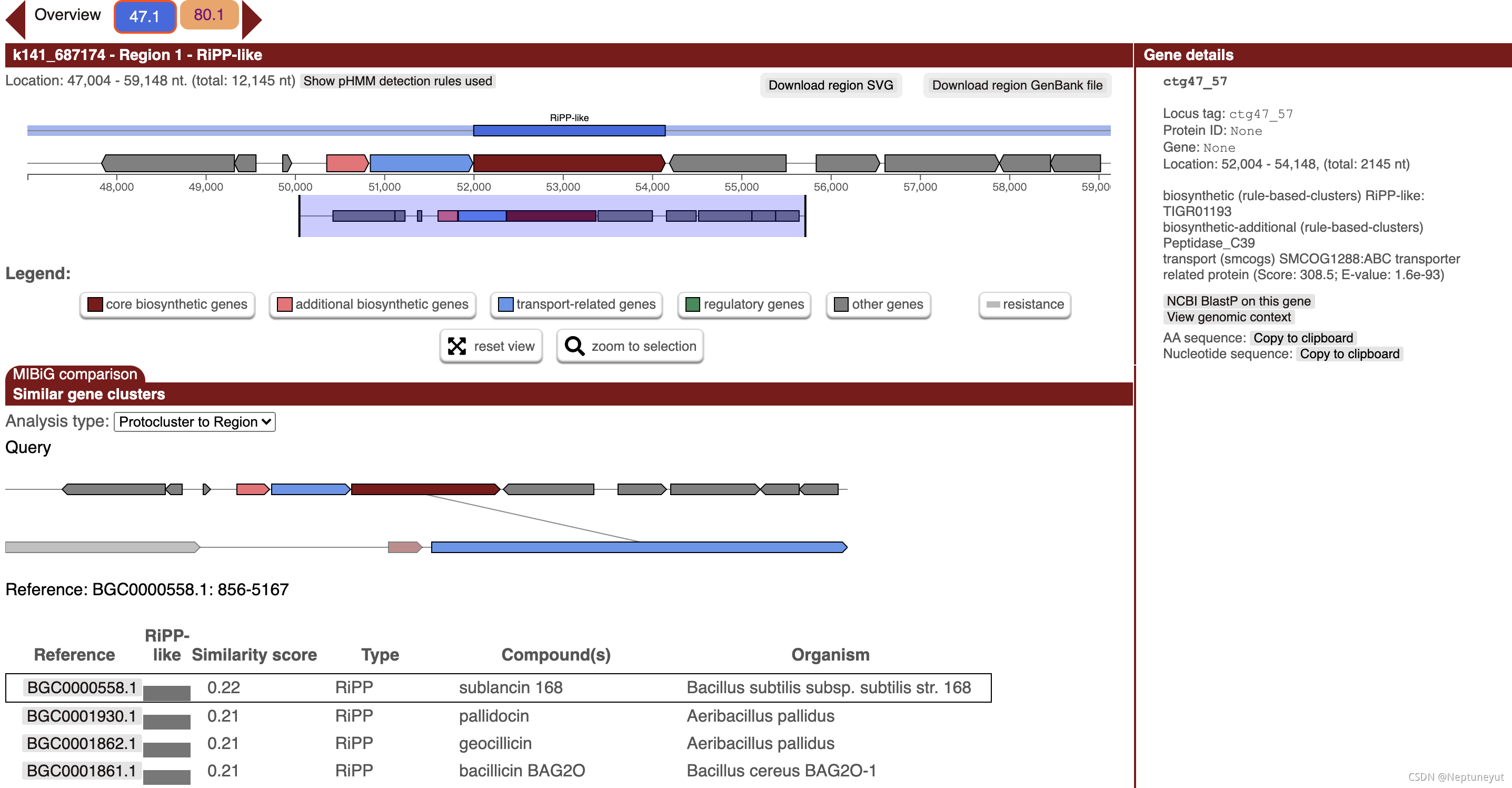 其他参数
# antiSMASH 6.0.1
--taxon bacteria --cb-general --cb-subclusters --cb-knownclusters --minlength 3000 --hmmdetection-strictness relaxed --genefinding-tool prodigal-m --smcog-trees --clusterhmmer --asf --smcog-trees --pfam2go --allow-long-headers --output-dir . --output-basename test
# --taxon {bacteria,fungi} Taxonomic classification of input sequence. (default: bacteria)
# --cb-general Compare identified clusters against a database of antiSMASH-predicted clusters.
# --cb-subclusters Compare identified clusters against known subclusters responsible for synthesising precursors.
# --cb-knownclusters Compare identified clusters against known gene clusters from the MIBiG database.
# --smcog-trees Generate phylogenetic trees of sec. met. cluster orthologous groups.
# --asf Run active site finder analysis.
# --pfam2go Run Pfam to Gene Ontology mapping module.
# --output-dir OUTPUT_DIR Drectory to write results to.
# --minlength MINLENGTH Only process sequences larger than (default: 1000).
# --allow-long-headers Prevents long headers from being renamed
# --output-basename OUTPUT_BASENAME Base filename to use for output files within the output directory.
# --hmmdetection-strictness {strict,relaxed,loose} Defines which level of strictness to use for HMM-based cluster detection, (default: relaxed).
输出详细结果
网络文件
其他参数
# antiSMASH 6.0.1
--taxon bacteria --cb-general --cb-subclusters --cb-knownclusters --minlength 3000 --hmmdetection-strictness relaxed --genefinding-tool prodigal-m --smcog-trees --clusterhmmer --asf --smcog-trees --pfam2go --allow-long-headers --output-dir . --output-basename test
# --taxon {bacteria,fungi} Taxonomic classification of input sequence. (default: bacteria)
# --cb-general Compare identified clusters against a database of antiSMASH-predicted clusters.
# --cb-subclusters Compare identified clusters against known subclusters responsible for synthesising precursors.
# --cb-knownclusters Compare identified clusters against known gene clusters from the MIBiG database.
# --smcog-trees Generate phylogenetic trees of sec. met. cluster orthologous groups.
# --asf Run active site finder analysis.
# --pfam2go Run Pfam to Gene Ontology mapping module.
# --output-dir OUTPUT_DIR Drectory to write results to.
# --minlength MINLENGTH Only process sequences larger than (default: 1000).
# --allow-long-headers Prevents long headers from being renamed
# --output-basename OUTPUT_BASENAME Base filename to use for output files within the output directory.
# --hmmdetection-strictness {strict,relaxed,loose} Defines which level of strictness to use for HMM-based cluster detection, (default: relaxed).
输出详细结果
网络文件
每次运行都会产生自己的一套输出文件,可以使用其他工具进行分析(如Cytoscape)。 Network_Annotations_Full.tsv 一个以制表符分隔的 |
【本文地址】
今日新闻 |
推荐新闻 |